ACG Clinical Guideline: Genetic Testing and Management of He… : Official journal of the American College of Gastroenterology

Hereditary gastrointestinal (GI) cancer syndromes represent a phenotypically diverse group of disorders that exhibit distinct patterns of inheritance in an individual’s progeny. Over the past few decades, the expansion of familial cancer registries and advancement in genomics have led to the development of clinical diagnostic criteria for specific hereditary syndromes as well as the discovery of multiple genes in which germline mutations predispose individuals to syndrome-associated neoplastic manifestations. This guideline first discusses essential elements of a patient’s personal and family history that allow for risk assessment for potential inherited cancer susceptibility. It then addresses the currently most well-characterized GI cancer susceptibility syndromes: Lynch syndrome (LS), familial adenomatous polyposis (FAP), attenuated familial adenomatous polyposis (AFAP), MUTYH-associated polyposis (MAP), Peutz–Jeghers syndrome (PJS), juvenile polyposis syndrome (JPS), Cowden syndrome (CS), serrated (hyperplastic) polyposis syndrome, hereditary pancreatic cancer, and hereditary gastric cancer. For each of these syndromes, we outline diagnostic criteria and indications for genetic evaluation, describe the currently known associated underlying genes, and make recommendations for surveillance and management of at-risk individuals and those found to carry a definitive disease-causing mutation. Finally, we discuss the elements of informed consent that must accompany genetic evaluation as well as currently evolving genetic testing technologies that may change how genetic testing is conducted in the near-term future.
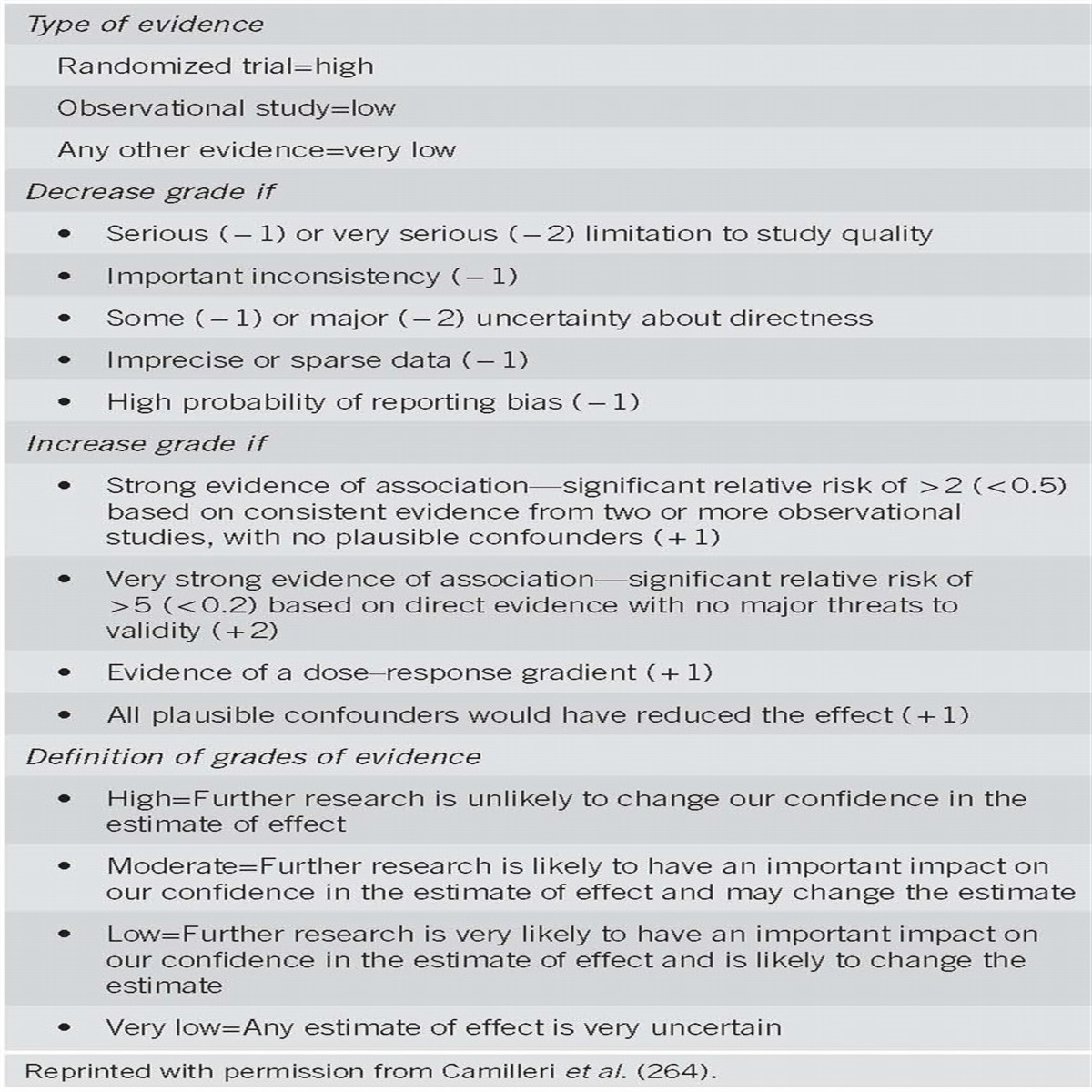
Each section of the document presents summary statements, the key recommendations related to the section topic, followed by a summary of the supporting evidence (Tables 1 and 2). A search of MEDLINE via the OVID interface using the MeSH term “hereditary cancer syndrome” limited to clinical trials, reviews, guidelines, and meta-analysis for the years 1966–2013 was performed to develop the document and create summary statements and recommendations. “Summary statements” and “recommendations” are distinguished by whether it was possible to address the quality of evidence supporting the statements based on an objective grading system. An objective measure that provides assessment of the strength of data regarding prognostic indicators does not currently exist, and similarly, “motherhood” statements (such as the importance of obtaining a family history) that are based on sound clinical judgment are often not subject to systematic clinical studies as they are understood to reflect sound clinical practice. The summary statements therefore reflect consensus opinion by the authors and a thorough literature review that reflects expert opinion by leaders in the field and other consensus guidelines. For management recommendations, where alternative strategies are and should be subject to rigorous assessment, the GRADE (Grading of Recommendations Assessment, Development and Evaluation) system was used to grade the strength of recommendations and the quality of evidence (1). An explanation of the quality of evidence and strength of recommendations is shown in Table 3. The quality of evidence, which influences the strength of the recommendation, ranges from “high” (further research is very unlikely to change our confidence in the estimate of effect) to “moderate” (further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate) to “low” (further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate), and to “very low” (any estimate of effect is uncertain). The strength of a recommendation is graded as strong when the desirable effects of an intervention clearly outweigh the undesirable effects and is graded as conditional when uncertainty exists about the trade-offs.

Summary statements

Summary of recommendations

Continued.

GRADE (Grading of Recommendations Assessment, Development and Evaluation) system of evidence and strength of recommendation
The field of cancer genetics poses some challenges with respect to the GRADE system. Because of the rarity of the syndromes, and the relatively recent discovery of cancer susceptibility genes, data regarding long-term outcomes regarding optimal management strategies at this time are limited to observational studies. Randomized clinical trials, which are the gold standard of systems such as GRADE, are difficult to conduct in rare diseases, where the main objective outcome, reduction in cancer mortality, takes years to assess and large patient numbers. The reader, therefore, should take the assessments of quality of evidence with caution—the often “low” or “very low” quality gradings reflect primarily a lack of available data and not that the quality of studies conducted thus far has been poor.
STANDARDS FOR MINIMAL CANCER FAMILY HISTORY ASSESSMENT IN GI PRACTICE
Summary statements
- A family history of cancer and premalignant GI conditions that provides sufficient information to develop a preliminary determination of the risk of a familial predisposition to cancer should be obtained for all patients being evaluated in outpatient gastroenterology and endoscopy practices.
- Essential elements of a family history include presence and type of cancer diagnoses in first- and second-degree relatives, and presence and (ideally) type of polyps in first-degree relatives; age and lineage should be noted for each diagnosis.
Summary of evidence
Approximately 5–10% of cancers are attributable to a hereditary cancer predisposition syndrome. Identifying those patients who have an inherited cancer predisposition syndrome has significant benefit to both the patient and at-risk relatives. For the index patient, the diagnosis of a hereditary cancer syndrome has implications for his/her surveillance strategy for multiple component tumors in terms of age of initiation and intervals between surveillance exams, and may lead to the consideration of prophylactic surgery or more extensive surgery in the case of neoplasia development. The diagnosis of an inherited syndrome also has significant implications for management of the patient’s immediate and extended family.
Features of a patient’s personal history may be the initial clue to the possibility of an inherited predisposition to cancer. Hallmark features, whose specifics are outlined in detail in the remainder of this guideline, include early age at onset of polyps or cancer and unusual numbers or histologies of cancers or premalignant conditions. Family history is the other key component to the identification of those individuals who may have an inherited predisposition to malignancy or who are at increased risk for additional primary cancers.
The goal of any cancer family history, in combination with the patient’s personal history, is to provide enough information to make a preliminary determination about whether the patient may have a familial predisposition to cancer, may benefit from genetic counseling and possibly testing for underlying cancer susceptibility genes, or may not need genetic counseling and testing, but still require more intensive surveillance than the average- or moderate-risk patient. As there is currently no clear evidence base to define how family history should be taken or what constitutes the right amount of information for an initial cancer screening family history, an expert panel was recently convened to define how and what to collect for a family cancer history (2). The panel agreed that although the gold standard family history is the comprehensive, three-generation pedigree used in medical genetics, counseling, and research settings, this evaluation is time consuming and not feasible in general medical practice. For most patients, family history of cancer and premalignant conditions in close relatives is most relevant. Guidelines for consideration of genetic risk assessment, such as the NCCN Clinical Guidelines in Oncology for Genetic/Familial High-Risk Assessment: Breast and Ovarian, (3) focus on first- and second-degree relatives, although they may optionally incorporate family history in third-degree relatives. Thus, family history of cancer in first-degree (parents, children, and siblings) and second-degree (grandparents, aunts/uncles, nieces/nephews, grandchildren, and half-siblings) relatives is often sufficient to assess a patient’s empiric risk of common cancers or a cancer patient’s risk of a second primary cancer. Relatives’ age at cancer or polyp diagnosis should also be assessed because this factors into both genetic risk assessment guidelines and cancer screening recommendations. Maternal and paternal lineages should be assessed separately. Accuracy of self-reported cancer family history in first-degree relatives (FDRs) has been shown to be >75% for most cancers, including colorectal, breast, ovarian, and pancreatic cancers. Studies have shown a decrease in the accuracy of reported family history in more distant relatives, ranging from 50 to 80% depending on the cancer (4, 5). Hence, the routine review of family medical records, although not required during family history collection, can be helpful in particular cases where the cancer site is in question.
LYNCH SYNDROME (LS)
Tumor testing and indications for genetic testing
Summary statements
- All newly diagnosed colorectal cancers (CRCs) should be evaluated for mismatch repair deficiency.
- Analysis may be done by immunohistochemical testing for the MLH1/MSH2/MSH6/PMS2 proteins and/or testing for microsatellite instability (MSI). Tumors that demonstrate loss of MLH1 should undergo BRAF testing or analysis for MLH1 promoter hypermethylation.
- Individuals who have a personal history of a tumor showing evidence of mismatch repair deficiency (and no demonstrated BRAF mutation or hypermethylation of MLH1), a known family mutation associated with LS, or a risk of ≥5% chance of LS based on risk prediction models should undergo genetic evaluation for LS.
Summary of evidence
LS, the most common cause of inherited CRC, is an autosomal-dominant condition defined by the presence of a germline mutation in a DNA mismatch repair gene (or EPCAM). It was often previously referred to as hereditary nonpolyposis colorectal cancer. LS tumors are associated with changes in the length of nucleotide repeat sequences of tumor DNA, termed MSI. MSI results from defective mismatch repair and is associated with loss of expression of the MLH1, MSH2, MSH6, and/or PMS2 proteins that can be detected by immunohistochemical (IHC) analysis. Multiple international studies have demonstrated that the prevalence of MSI in population-based series of CRC ranges from 7 to 19% (6, 7, 8, 9, 10). The sensitivity of MSI testing among those with MLH1 or MSH2 mutations is 80–91%, and is 55–77% among those with MSH6 or PMS2 mutations; the specificity of MSI testing is 90% (11). The sensitivity of IHC testing, regardless of the MMR gene involved, is 83% and the specificity is 89% (11). MSI and IHC results are highly correlated (9, 12), and as protein staining is often easier to perform than DNA analysis in a clinical setting, it may be a more feasible option for widespread MSI screening. In order to facilitate surgical planning, tumor testing on suspected CRC should be performed on preoperative biopsy specimens if possible. For individuals whose IHC indicates loss of the MLH1 protein, determination of the mechanism of loss should be pursued as an additional screening step, and this may be done by analysis for a BRAF mutation or promoter hypermethylation studies. Almost no LS tumors carry a BRAF mutation, whereas 68% of those without LS do (11). Individuals who demonstrate evidence of MMR deficiency, independent of somatic MLH1 silencing, should undergo genetic testing.
Genetic etiology
Summary statement
- Genetic testing of patients with suspected LS should include germline mutation genetic testing for the MLH1, MSH2, MSH6, PMS2, and/or EPCAM genes (13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23), or the altered gene(s) indicated by IHC testing.
Summary of evidence
In 1993, genome-wide linkage analysis in several large families with autosomal-dominant CRC and the demonstration of associated tumor MSI led to the subsequent cloning of the mismatch repair genes MLH1 and MSH2, followed by MSH6, PMS2, and EPCAM (13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23). Multiple large international population-based series have demonstrated that MMR gene mutations account for 1–3% of newly diagnosed CRC cases (6, 7, 9, 10, 12). LS should be considered in individuals whose tumors show evidence of MMR deficiency as discussed above (without the presence of a BRAF mutation or MLH1 promoter hypermethylation), and those whose personal and/or family history fullfill the Amsterdam criteria, Bethesda Guidelines, or who have a ≥5% risk of carrying a germline mutation based on available prediction models (24, 25)(Table 4). The computational models all appear to outperform existing clinical guidelines (25), primarily because of limited sensitivity of the clinical criteria in identifying mutation carriers. In families where LS is a consideration, and no tumor sample is available for analysis, direct germline testing of an unaffected at-risk individual whose risk is calculated to be ≥5% based on the PREMM1,2,6 risk prediction model (accessible at http://premm.dfci.harvard.edu/) is a strategy that has been demonstrated to be cost effective in improving health outcomes (26). The complexity of clinical criteria may be difficult to apply in clinical practice. A simple, validated three-question tool may be used as a quick initial screen in busy practices to identify which patients need further risk assessment (27) (Table 4).

Amsterdam criteria, revised Bethesda guidelines, and colorectal cancer risk assessment tool
Surveillance and management of CRC
Recommendation
1. In individuals at risk for or affected with LS, screening for CRC by colonoscopy should be performed at least every 2 years, beginning between ages 20 and 25 years. Annual colonoscopy should be considered in confirmed mutation carriers (strong recommendation, moderate quality of evidence for screening, and very low quality of evidence for annual surveillance and age of initiation).
Summary of evidence
The precursor lesion for a LS-related CRC is an adenomatous polyp that is often proximal and can occasionally be flat rather than elevated/polypoid and likely to demonstrate villous features, high-grade dysplasia, and a preponderance of tumor-infiltrating lymphocytes (28). The historical term nonpolyposis CRC was intended to differentiate this condition from FAP in which patients develop hundreds of adenomas. LS patients develop few (usually <10) early-onset adenomas, but the adenoma–carcinoma sequence appears to be accelerated in LS with polyp to cancer dwell times estimated at 35 months compared with 10–15 years in sporadic cancer (29). Reports of lifetime risks of CRC for MLH1 and MSH2 gene mutation carriers range from 22 to 74% (30, 31, 32, 33, 34, 35) (Table 5). Lower risk for colorectal malignancy has been found in women, but not in men with MSH6 mutations (30% vs. 69% cumulative risk by age 70 years, respectively), compared with MLH1 and MSH2 carriers (36). CRC risk is reported to be lower in one study of 99 PMS2 mutation carriers, with an estimated cumulative risk of 15–20% by age 70 years. (37). The mean age of CRC diagnosis in LS patients is 44–61 years (8, 12, 38, 39) compared with 69 years in sporadic cases of CRC (40).

Cumulative risks of colorectal cancer in hereditary colorectal cancer syndromes
CRC prevention in LS families is guided by the distinctive characteristics of these malignancies, including the younger age of presentation, right-sided colorectal predominance, and rapid polyp growth with shorter dwell time before malignant conversion. Evidence for the effectiveness of colorectal screening in decreasing CRC mortality has been documented in studies by Järvinen et al. (41, 42, 43) (Table 6). Individuals at risk for LS who took up colonoscopic surveillance had 65% (P=0.003) less death from CRC compared with those who refused surveillance. Update of this Finnish study that analyzed colonoscopic surveillance in LS mutation carriers found no difference in CRC death between mutation carriers and mutation-negative relatives (43). Dove-Edwin et al. (44) reported the results of a prospective observational study of colonoscopy surveillance of members in hereditary nonpolyposis colorectal cancer or LS families, revealing a 72% decrease in mortality from CRC in those undergoing screening. In several studies (45, 46, 47, 48), more frequent colonoscopy screening (≤2 years) was associated with an earlier stage of CRC at diagnosis and less CRC than less frequent colonoscopy. At least every 2-year colonoscopic surveillance of LS patients is supported by the data above and the rapid adenoma–carcinoma sequence reported in these patients. In surveillance of MMR germline mutation-positive patients, consideration should be given to annual colonoscopy, as several studies have demonstrated CRC development with surveillance intervals that are between 1 and 2 years (29, 47). In carriers of deleterious MSH6 and PMS2 mutations, the risk of CRC is less and age of diagnosis later (37, 49) than in patients with MLH1 and MSH2 mutations. In these affected individuals, consideration could be given to starting surveillance at age 25–30 in MSH6 and PMS2 carriers (24), unless an early-onset cancer exists in a given family; however, this approach of gene-specific alterations in surveillance has not been evaluated in clinical studies.

Studies of colorectal screening in hereditary colorectal cancer (CRC) syndromes

Continued.
Recommendation
2. Colectomy with ileorectal anastomosis (IRA) is the preferred treatment of patients affected with LS with colon cancer or colonic neoplasia not controllable by endoscopy. Segmental colectomy is an option in patients unsuitable for total colectomy if regular postoperative surveillance is conducted (conditional recommendation, moderate quality of evidence).
Summary of evidence
The treatment for patients with CRC or premalignant polyps that cannot be removed by colonoscopy is subtotal colectomy with IRA. A high rate of metachronous CRC (16% at 10 years; 41% at 20 years) is noted in LS patients who have undergone segmental surgical resection of the initial CRC in several retrospective studies (48, 50, 51). A standard low anterior resection or abdominal perineal resection may be performed to treat rectal cancers in LS patients, although the residual colon is at high risk of metachronous neoplasia. Younger patients may be offered a total proctocolectomy and ileal pouch anal anastomosis (IPAA), or ileostomy. A recent retrospective study of 79 LS patients with rectal cancer who had undergone proctectomy found a cumulative risk of metachronous colon cancer to be 19% at 10 years, 47% at 20 years, and 69% at 30 years after surgical resection (51). The risk of metachronous cancer is substantially abated if extensive colectomy is performed (0–3.4%) (48, 50, 51).
In a Dutch study, no difference in global quality of life was noted between 51 LS patients who underwent partial colectomy and 53 patients who underwent subtotal colectomy, although functional outcome (stool frequency, stool-related aspects, social impact) was worse after subtotal colectomy than after partial colectomy (52). A comparison of life expectancy gained performing total colectomy vs. hemicolectomy in LS patients at ages 27, 47, and 67 years by Markov modeling was 2.3, 1, and 0.3 years, respectively. These investigators concluded that total colectomy is the preferred treatment in LS but hemicolectomy may be an option in older individuals. Consideration for less extensive surgery may be given in patients who are >60–65 years of age.
The option of prophylactic colectomy should be discussed with mutation carriers who have an endoscopically normal colon as an alternative to surveillance. Although rarely chosen, it may be attractive to patients from families where the prevalence of colon cancer is very high, or for whom colonoscopy is difficult. Direct comparative studies of extensive surgery vs. annual or biennial surveillance have not been conducted, and are unlikely be instituted because of the multitude patient-dependent factors that affect comorbidities and quality of life.
Surveillance and management of extracolonic malignancies
Gynecologic malignancies
Recommendations
3. Hysterectomy and bilateral salpingo-oophorectomy should be offered to women who are known LS mutation carriers and who have finished child bearing, optimally at age 40–45 years (conditional recommendation, low quality of evidence).
4. Screening for endometrial cancer (EC) and ovarian cancer should be offered to women at risk for or affected with LS by endometrial biopsy and transvaginal ultrasound annually, starting at age 30 to 35 years before undergoing surgery or if surgery is deferred (conditional recommendation, very low quality of evidence).
Summary of evidence
EC is the second most common cancer occurring in LS. Estimates of the cumulative lifetime risk of EC in LS patients range from 15 to 71%, with variability depending on specific gene mutation (31, 33, 35, 36, 37, 49) (Table 7); reports of age at diagnosis of this malignancy are clearly a decade or more younger than sporadic EC but range from 48 to 54 years (31, 33, 35, 36, 37, 49)(Table 7). Estimates of the cumulative lifetime risk of ovarian cancer in LS patients ranges from 3.4 to 22% (31, 38, 53, 54, 55) (Table 7).

Cumulative risks of extracolorectal cancer in hereditary colorectal cancer syndromes

Continued.
Because of the worrisome cumulative risk of EC, several annual screening modalities have been proposed including pelvic exams, transvaginal ultrasound, endometrial sampling, and CA 125 testing. Few studies of these interventions have been conducted and there is currently no evidence of survival benefit from EC surveillance (Table 8). Decrease in death from EC screening may be difficult to prove as 75% of LS patients with EC present with stage 1 disease and have an 88% 5-year survival rate. Transvaginal ultrasound has poor sensitivity and specificity for the diagnosis of EC in this population (56, 57). However, endometrial sampling appears useful in identifying some asymptomatic patients with EC and those with premalignant endometrial lesions (Table 8). Currently, no studies on the effectiveness of ovarian screening are available for women in LS families.

Studies of extracolonic cancer screening in hereditary colorectal cancer syndromes

Continued.
One retrospective study of 315 women with MMR mutations who did and did not have hysterectomy and oophorectomy revealed no cancers in the surgical group compared with a 33% and 5.5% rate of uterine and ovarian cancer, respectively, in the nonsurgical group (58). Cost-effectiveness analysis modeling of gynecological screening vs. prophylactic gynecological surgery (hysterectomy and bilateral salpingo-oophorectomy) in a theoretical population of 30-year-old women with LS revealed that prophylactic surgery had lower cost and higher quality-adjusted life-years (59). An additional modeling study evaluated multiple screening and surgical strategies. This investigation concluded that annual screening starting at age 30 years followed by prophylactic surgery at age 40 years was the most effective gynecologic cancer prevention strategy, but incremental benefit over prophylactic surgery at age 40 years alone was attained at substantial cost (60).
Recommendations
5. Screening for gastric and duodenal cancer can be considered in individuals at risk for or affected with LS by baseline esophagogastroduodenoscopy (EGD) with gastric biopsy at age 30–35 years, and treatment of Helicobacter pylori infection when found. Data for ongoing regular surveillance are limited, but ongoing surveillance every 3–5 years may be considered if there is a family history of gastric or duodenal cancer (conditional recommendation, very low quality of evidence).
6. Screening beyond population-based recommendations for cancers of the urinary tract, pancreas, prostate, and breast is not recommended unless there is a family history of the specific cancers (conditional recommendation, low quality of evidence).
Summary of evidence
The impact of a family history of extracolonic cancers on other at-risk relatives has not been systematically studied. Some studies show clustering of extracolonic cancers in families, whereas others have not (discussed when available in section below for individual cancers). In clinical practice, decision making regarding surveillance for extracolonic cancers is generally done on a case-by-case basis, taking into account cancer history in at-risk first- and second-degree relatives on the affected side of the family.
Some studies have estimated the lifetime risk of gastric cancer in LS to be as high as 13%, but it is currently much lower in North America and Western Europe. A carefully conducted time trend study of gastric cancer found an 8.0% and 5.3% lifetime risk of this malignancy in males and females with MMR gene mutation, respectively, and lack of familial clustering (61). The majority of gastric cancers in LS patients appear to be histologically classified as intestinal type (61, 62), and, consequently, potentially amenable to endoscopic surveillance. There are no studies that have evaluated the effectiveness of screening and surveillance for gastric cancer in LS patients.
The lifetime risk for small bowel cancer ranges from 0.4 to 12.0% (31, 34, 53, 54, 63, 64). The majority of small bowel cancers in a LS cohort were located in the duodenum or ileum (65) and within the reach of EGD and colonoscopy with dedicated ileal intubation. Studies of small bowel screening in LS patients are lacking. However, one screening investigation of 35 gene mutation carriers found that 2 had jejunal adenomas and 1 had a jejunal cancer (66) (Table 8). Six additional patients had capsule endoscopy images of uncertain clinic relevance, prompting further invasive investigation in five patients. A recent publication suggested that routine surveillance of the small bowel in LS was not cost efficient (55).
Estimates of the lifetime risk of urinary tract cancer in LS range from 0.2 to 25%, depending on the study and which urinary tract cancers are included (transitional cell carcinoma of the ureter, renal pelvis, and bladder) (31, 53, 54, 63, 64, 67). Currently, a dearth of literature on screening for urinary cancer in LS patients exists. One retrospective study evaluating screening for urinary cancer by urine cytology in individuals in hereditary nonpolyposis colorectal cancer or LS families found a poor (29%) sensitivity in diagnosing cancer in asymptomatic patients and production of many false positive results requiring invasive investigation (68) (Table 8). Screening has not been shown to be effective with urine cytology and urinalysis for microscopic hematuria for urinary cancer in the general population and in groups at higher risk for bladder cancer from environmental factors (69, 70). The benefit of ultrasound screening is unknown. In summary, limited data exist to advocate urinary screening.
The risk of pancreatic cancer (PC) in LS patients was noted to be elevated in two cohort studies. In one study, the standardized incidence ratio for PC was 10.7 (95% confidence interval, 2.7–47.7), with a 10-year cumulative risk of 0.95% (71), and the other study reported a 8.6-fold increase (95% confidence interval, 4.7–15.7), with cumulative risk of 3.7% by age 70 years (72). The benefit of screening for PC in LS has not been evaluated. An international pancreas consensus panel recommended that, based on expert opinion, mismatch repair gene mutation carriers with one affected FDR should be considered for annual PC surveillance with magnetic resonance imaging (MRI) and/or endoscopic ultrasound based on early data in other cohorts with comparable risk (73).
There are conflicting data regarding the risk of several other extracolonic cancers in patients with LS. The relationship between LS and breast cancer is unclear. Although a small increase in lifetime breast cancer risk of 18% has been found (54, 74), most clinic-based registry reports have not demonstrated this consistently (55, 75). In two recent studies the relative risk of prostate cancer is 2.0- to 2.5-fold the general population risk (54, 76); however, the effectiveness of intensive screening beyond population recommendations has not been evaluated.
Prevention strategies
Diet, exercise, smoking, and supplements. A prospective analysis of 386 patients with LS undergoing surveillance revealed that current smokers had an increased risk of colorectal adenomas compared with past smokers and never smokers (hazard ratio of 6.1 vs. 3.0 vs. 1, respectively (77). Excess body weight (body mass index >25 kg/m2) has been shown to be associated with an elevated risk (hazard ratio of 8.7 compared with normal weight) of colorectal adenomas in men with LS in the same cohort; an elevated risk was not found in women with a high body mass index (78).
Chemoprevention
Recommendation
7. Although data suggest that daily aspirin may decrease the risk of colorectal and extracolonic cancer in LS, currently the evidence is not sufficiently robust or mature to make a recommendation for its standard use (conditional recommendation, moderate quality of evidence).
Summary of evidence
Resistant starch and aspirin have been assessed as chemopreventive agents in patients with LS. The Colorectal/Adenoma/Carcinoma Prevention Programme 2 (CAPP2) was a randomized placebo-controlled trial with a two-by-two design investigating the effect of resistant starch (Novelose) 30 g per day and aspirin 600 mg per day taken for up to 4 years on development of colorectal adenoma and cancer (79). This study randomized 727 participants to starch or placebo and 693 between aspirin and placebo. The use of resistant starch, aspirin, or both had no effect on the incidence of colorectal neoplasia in LS carriers over a mean follow-up period of 29 months. The CAPP2 investigators subsequently evaluated the long-term effect of 600 mg of aspirin usage on CRC development (80). At a mean follow-up of 55.7 months, intention-to-treat analysis of time to first CRC showed a hazard ratio of 0.63 (95% confidence interval, 0.35–1.13, P=0.12). An intention-to-treat analysis of all LS cancers (colorectal, endometrial, ovarian, pancreatic, small bowel, gallbladder, ureter, stomach, kidney, and brain) revealed a protective effect of aspirin vs. placebo (hazard ratio, 0.65; 95% confidence interval, 0.42–1.00, P=0.05). During the intervention, adverse events did not differ between aspirin and placebo groups.
The CAPP2 trial has several limitations. First, ascertainment of the end point, CRC, was not standardized, and more intensive colonoscopic evaluation could have occurred in the aspirin group than in the non-aspirin group because of more frequent adverse effects after intervention. Second, the extracolonic cancers did not undergo molecular evaluation to assess whether they were related to the germline MMR mutation. In addition, the dose of daily aspirin utilized in the CAPP2 trial is significantly higher than that noted to be effective (75 mg a day) in sporadic CRC chemoprevention.
The CAPP3 trial is currently underway to establish the optimum dose and duration of aspirin treatment. Although data exist to suggest that aspirin may decrease the risk of colorectal and extracolonic cancer in LS, currently the evidence is not sufficiently robust or mature to make a recommendation for its standard use.
Adenomatous polyposis syndromes
Familial adenomatous polyposis/MUTYH-associated polyposis/attenuated polyposis indications for genetic evaluation
Summary statement
- Individuals who have a personal history of >10 cumulative colorectal adenomas, a family history of one of the adenomatous polyposis syndromes, or a history of adenomas and FAP-type extracolonic manifestations (duodenal/ampullary adenomas, desmoid tumors (abdominal>peripheral), papillary thyroid cancer, congenital hypertrophy of the retinal pigment epithelium, epidermal cysts, osteomas) should undergo assessment for the adenomatous polyposis syndromes.
Summary of evidence
There are three known hereditary syndromes where inheritance of germline mutations produces enhanced colorectal carcinogenesis, manifested by early age of onset of multiple colorectal adenomas with the potential for early development of CRC: FAP, AFAP and MAP.
FAP is the defined by the presence of ≥100 synchronous colorectal adenomas inherited in an autosomal-dominant manner. Estimates of the prevalence of FAP vary from 1 in 6,850 to 1 in 31,250 live births (2.29 to 3.2 cases per 100,000 individuals) (81, 82, 83, 84, 85, 86, 87). The frequency is fairly constant throughout the world, with men and women being affected equally.
Patients with 10 to 99 synchronous adenomas have oligopolyposis or AFAP. AFAP is defined by <100 adenomas at presentation inherited in an autosomal-dominant pattern. Patients with AFAP have fewer adenomas than those with typical FAP, averaging 25 polyps in one study and exhibiting a more proximal colonic preponderance than in typical FAP (88, 89, 90). However, the polyp number is extremely variable within many kindreds.
In 2002, an attenuated polyposis syndrome was described in three siblings affected with multiple adenomas and/or CRC inherited in an autosomal-recessive pattern (91). This recessive condition is referred to as MAP and is characterized by an increased risk for CRC and multiple adenomatous polyps that can mimic FAP or AFAP.
There are two settings in which to consider genetic testing for the adenomatous polyposes syndromes: (i) testing an individual with a phenotype suggestive of one of the polyposes, but where the clinical diagnosis is not certain, and (ii) testing relatives of a patient with a known germline mutation. The first setting is usually defined as a patient with ≥10 cumulative adenomas, or sometimes suggestive extracolonic manifestations, but no known family history of an underlying pathogenic mutation. Genetic testing in this setting should be comprehensive and the absence of a mutation does not definitively rule out a clinical diagnosis if the phenotype is striking. In the second setting, relatives of an individual with a known pathogenic mutation are tested for the presence or absence of that particular mutation. A positive test indicates the diagnosis of a syndrome, whereas a negative test (absence of mutation) rules it out and establishes that the individual is not at a syndromic risk for cancer and polyps.
Genetic etiology
Summary statement
- Genetic testing of patients with suspected adenomatous polyposis syndromes should include APC and MUTYH gene mutation analysis.
Summary of evidence
FAP arises from germline mutations of the APC gene on chromosome 5q21 (92, 93). FAP is dominantly inherited and is close to 100% penetrant. Up to one-third of newly diagnosed cases not belonging to previously identified families appear to represent either de novo germline mutations or mosaicism (81, 94, 95, 96, 97, 98).
APC is a tumor suppressor gene; thus, gene inactivation occurs only after both alleles are mutationally damaged. In FAP, one allele is inherited in a mutated form. Adenoma formation is initiated when the second allele is damaged or lost by a somatic event. The progression of adenoma to carcinoma after APC inactivation is similar in FAP and the sporadic setting in that mutations accumulate in additional relevant genes including K-ras, p53, a gene or genes on chromosome 18, and possibly others (99). Although mutations have been found scattered throughout the APC gene, most are located in the 5Í´ end of exon 15, often called the mutation cluster region (100, 101). The location of mutations in the APC gene correlate to some degree with colonic adenoma number, desmoid tumor occurrence, and congenital hypertrophy of the retinal pigment epithelium (102). Individuals with >1,000 polyps exhibit mutations in the mid-portion of the gene (102, 103).
AFAP arises from APC mutations at either the far proximal (5Í´) end of the gene, the far distal (3Í´) end of the gene, or in certain locations of exon 9 (90, 102). Whole or partial gene deletions may also give an attenuated phenotype.
MAP is a recessively inherited syndrome due to biallelic (homozygous or compound heterozygous) MUTYH mutations. MUTYH is a base excision repair gene involved in DNA oxidative damage repair whose protein repairs oxidative damage to the DNA (91). Failure of base excision repair results in CG–AT transversions in multiple genes, including APC and KRAS (91, 104, 105). Polyp and cancer predisposition occur with germline MUTYH mutations, but somatic MUTYH mutations do not appear to play a role in the pathogenesis of colon cancer (106, 107). The two most prevalent MUTYH mutations, occurring in >80% of individuals of European ancestry with MAP, are two missense mutations Y179C and G396D (previously referred to as Y165C and G382D, respectively) (91, 104, 106, 108, 109, 110). Other population-specific MUTYH mutations have been found (107). Approximately 90% of “western” population MAP patients have at least one of these two mutations; however, many other distinct MUTYH mutations have been reported (107).
MAP is most commonly found in patients presenting with 20 to 99 adenomas (107, 111). Biallelic MUTYH mutations are found in 7.5% to 12.5% of patients with >100 adenomas in whom a disease-causing APC mutation is not found (104, 112) and in 16 to 40% of patients with 15 to 99 colonic adenomas but not FAP (104, 108, 109, 113, 114). Although biallelic mutations have been found in individuals with early-onset CRC and few to no polyps, and in individuals with <10 adenomas without CRC, this is relatively uncommon (115). MAP was found in 0 out of 400 individuals with <4 adenomas, 2 of 444 (0.5%) unselected CRCs, and 0 out of 62 MSI high CRCs (112). Similar frequencies among patients with polyps and CRCs have been found in other studies. In a study from Finland, 0.4% of 1,042 population-based CRC cases had biallelic MUTYH mutations (110). All those with mutations were found to have adenomas, ranging from 3 to 100. In a large (2,239 cases and 1,845 controls) population-based case–control study from Scotland, 0.8% of CRC cases <55 years old and 0.54% of all cases had biallelic mutations (116).
Monoallelic MUTYH mutations are found in 1 to 2% of the general population (107). Monoallelic MUTYH mutation carriers may have a slightly elevated risk of CRC, although the precise magnitude of the increased risk is currently unclear; most studies estimate a 1.5–2-fold risk above the general population (107). There is currently no consensus regarding the management of monoallelic carriers as data are limited. An option for clinicians at the current time is to manage monoallelics as individuals with a FDR with CRC, offering colonoscopy as a surveillance modality every 5 years, beginning 10 years earlier than the earliest CRC diagnosis.
Polymerase-proofreading associated polyposis is a newly described syndrome and only a few families have been characterized (117). Its phenotype includes oligo-adenomatous polyposis and an early age of onset of colorectal and EC. In a recent study of 858 familial/early-onset CRC cases and polyposis, one known POLE germline mutation and one new POLD1 mutation were identified (118). Polymerase proofreading-associated polyposis is dominantly inherited and penetrance appears high.
Patients with clinical suspicion of an adenomatous polyposis syndrome should have genetic counseling and testing for germline mutations in APC and MUTYH. Failure to identify a mutation in an index case does not rule out the diagnosis of adenomatous polyposis, as mutations cannot be found in all families. If testing is negative, and clinical suspicion remains high, testing for other possible underlying genes should be considered. Failure to find a mutation means that all close relatives must still be screened as if they have FAP.
Finding a mutation confirms the diagnosis of adenomatous polyposis and allows relatives to be tested with a high degree of accuracy. Once an affected patient has been genotyped, all at-risk relatives can be screened for the mutation.
Surveillance and management of CRC and polyps
Recommendation
8. In individuals at risk for or affected with the classic AP syndromes, screening for CRC by annual colonoscopy or flexible sigmoidoscopy should be performed, beginning at puberty. In families with AFAP or MAP, surveillance should be by colonoscopy (strong recommendation, moderate quality of evidence).
Summary of evidence
Colon screening should be performed in those with a clinical or genetic diagnosis of FAP or in FDRs of those with FAP if genetic testing is uninformative or has not been done. In families where no mutation can be found, all at-risk relatives must undergo endoscopic screening. Colonoscopy should begin at puberty, or whenever there are suggestive symptoms such as chronic diarrhea, rectal bleeding, or abdominal pain. Flexible sigmoidoscopy is also reasonable in families with classic FAP, until a polyp is found. If this is proved to be an adenoma, full colonoscopy should be done. During colonoscopy, polyp number, size, and distribution should be recorded, and several polyps should be biopsied. The average age of FAP diagnosis in patients presenting with symptoms is 35.8 years (range, 4–72 years) in the St Marks FAP registry in London (81). Polyps begin to appear most often in the second or third decade of life. The mean age of polyp occurrence is 15.9 years (range, 8–34 years) (119). Adenomatous polyps are usually distributed evenly throughout the colon, with a slight distal colonic excess. The size of the polyps depends on the stage at which the patient is examined. Even in fully developed cases, however, 90% of adenomas are <0.5 cm in diameter, and <1% of polyps are >1 cm. Polyps may either carpet the colon with myriad small lesions or occur as more distinct and somewhat larger lesions. Striking heterogeneity of polyp number and growth rate has been observed (120). Histopathology demonstrates tubular adenomas, indistinguishable from common or sporadic adenomas. Villous and tubulovillous histologies are also seen, but much less frequently and usually in larger polyps. A histologic feature of FAP not observed in the general population is dysplastic or adenomatous epithelial cells in single crypts or even portions of single crypts. These are called microadenomas and are often seen in FAP biopsy specimens of normal-appearing mucosa (81). Budding of dysplastic epithelium from normal crypts can be observed and aberrant crypt foci have been reported to occur with increased frequency in FAP (121). These lesions are similar to microadenomas but are identified with methylene blue staining of the colonic mucosal surface.
Colonic adenocarcinoma is the inevitable consequence of FAP unless the colon is removed. There is a 25% incidence of colon cancer in newly diagnosed FAP patients, not belonging to known families, that remains common because of the high frequency of de novo germline mutations (94, 122). In the St Mark’s series, the average age at cancer diagnosis was 39 years. By 45 years of age, 87% had developed cancer, and by 50 years, it increased to 93%. Colon cancer has been reported as early as 9 years of age, although the occurrence of malignancy before adolescence is very unusual. Multiple colonic malignancies were present in ∼48% of those with cancer (41% synchronous and 7% metachronous). Of the malignancies, 84% were at or distal to the splenic flexure, a fraction almost identical to that found in their series of random colorectal malignancies at that time. Average life expectancy after diagnosis of cancer was 2.6 years.
The generally accepted colon screening guideline for children at risk for classic FAP is every 1- to 2-year sigmoidoscopy beginning at 10 to 12 years of age (123, 124, 125, 126, 127). Those initially screened at an older age should probably have colonoscopy for the first examination. If surgery is delayed longer than a year after polyps emerge, annual colonoscopy should be used for surveillance.
Colon screening with subsequent surgery decreases and almost eliminates mortality from large bowel malignancy in FAP (127, 128, 129, 130). Survival is remarkably improved in relatives of probands who undergo screening (82, 85, 86, 129, 131).
In AFAP, the emergence of adenomas and cancer is delayed 10 to 20 years compared with typical FAP. The cumulative risk of CRC in AFAP by age 80 years in two large carefully studied kindreds was estimated to be 69%, with an average age at cancer diagnosis of 58 years (range, 29–81 years) (90). In another study, the average age at symptomatic presentation was 52 years (132). For AFAP, colonoscopy should always be used for screening, in view of more proximal colonic polyp distribution. Onset of examination in AFAP can reasonably be delayed until the late teens to mid-20s and be performed every 1 to 2 years.
MAP patients commonly have between 20 and 99 polyps (107, 111) and rarely have >500, although the colonic phenotype can vary (107, 112, 113). CRC was found to be present in patients with an MAP diagnosis in ∼60% of cases (107). CRCs in MAP have been predominantly distal colonic in some studies (105) and proximal in others (133). In eight population-based studies, 28 of 79 (35%) MAP cases with CRC had no concurrent polyps, whereas 17 (22%) had <10 adenomas (107). The risk of CRC by age 50 years is 19% and by age 60 years is 43%, with an average age of onset of 48 years (107, 134, 135). Although the predominant polyp type in patients with MAP is an adenoma, multiple hyperplastic and/or sessile serrated polyps (also referred to as sessile serrated adenomas) may occur. In a small study of 17 patients with MAP, 8 (47%) had at least one hyperplastic and/or sessile serrated polyp, 3 (˜18%) met criteria for serrated polyposis (previously referred to as hyperplastic polyposis, see Serrated polyposis section below for additional details), and 1 patient had over 100 hyperplastic and sessile serrated polyps (136). Treatment of MAP follows the same principles as AFAP. The disease may be managed endoscopically with at least yearly colonoscopy. If the polyps become endoscopically uncontrollable, then colectomy is indicated. Currently, there is no consensus as to whether monoallelic MUTYH mutations warrant increased CRC screening.
No recommendations for treatment or surveillance of patients with polymerase proofreading-associated polyposis have been made because the frequency of polyps, cancer, and the extracolonic phenotype have yet to be determined, but the options of close endoscopic surveillance and colectomy seem reasonable.
Recommendation
9. Absolute indications for immediate colorectal surgery in FAP, AFAP, and MAP include: documented or suspected cancer or significant symptoms. Relative indications for surgery include the presence of multiple adenomas >6 mm, a significant increase in adenoma number, the presence of an adenoma with high-grade dysplasia, and inability to adequately survey the colon because of multiple diminutive polyps (strong recommendation, low quality of evidence).
Summary of evidence
Development of colon cancer in classic FAP is inevitable if the colon is not removed. An appropriately timed colectomy remains the cornerstone of colon cancer prevention in FAP (127, 137, 138). Prophylactic surgery can be planned at a suitable time (late teens to early twenties), based on the risk of cancer posed by the polyp burden. Indications for early surgery include polyps >10 mm diameter, polyps with high-grade dysplasia, marked increases in polyp number from one exam to the next, and symptoms. Surgical options are colectomy with IRA (for <20 rectal and <1,000 colonic adenomas) and proctocolectomy with IPAA (for severe or profuse adenomas, >20 rectal adenomas, and >1,000 colonic adenomas). A laparoscopic approach is now often used for both surgical approaches. Conversion from IRA to IPAA may occasionally be needed because of development of numerous or advanced rectal adenomas. Proctocolectomy with ileostomy is rarely needed. APC mutation location, allowing prediction of severity of rectal polyposis and likelihood of future completion proctectomy, has been suggested as a factor to consider in determining which procedure should be done (139).
Colectomy with IRA is a single-stage procedure with slightly less morbidity than the IPAA surgery, but some rectal cancer risk remains and yearly proctoscopy is essential (139, 140). Even after total proctocolectomy and IPAA, adenomas and cancers may occur in the anal transition zone and in the pouch itself; lifelong endoscopic surveillance is required (141, 142, 143). Possible morbidities from either surgery include increased bowel frequency and incontinence. Pouch surgery is associated with some loss of fertility in women and some loss of sexual function in men.
Patients with AFAP can often be managed for many years with colonoscopic polypectomy and may possibly never need colectomy (90). If surgical resection is indicated, AFAP patients can almost always undergo colectomy and IRA because of rectal sparing of polyps. After colectomy with IRA in a large series of patients with AFAP, an average of 3.4 recurrent polyps (range, 0–29) and only one cancer was found in the postcolectomy rectal remnant over a mean follow-up of 7.8 years (range, 1–34 years) (90).
Similar to AFAP, FAP, and some multiple adenoma patients, subtotal colectomy with close subsequent surveillance would seem to be the best option for MAP patients with relative rectal sparing. Restorative proctocolectomy is indicated if the rectum is substantially involved.
Surveillance and management of extracolonic malignancies
Recommendations
10. Screening for gastric and proximal small bowel tumors should be done using upper endoscopy including duodenoscopy starting at age 25–30 years. Surveillance should be repeated every 0.5–4 years depending on Spigelman stage of duodenal polyposis: 0=4 years; I=2–3 years, II=1–3 years, III=6–12 months, and IV=surgical evaluation. Examination of the stomach should include random sampling of fundic gland polyps. Low-grade dysplasia is common in fundic gland polyps, and surgery should be reserved for high-grade dysplasia or cancer (strong recommendation, very low quality of evidence).
11. Annual thyroid screening by ultrasound should be recommended to individuals affected with FAP, MAP, and attenuated polyposis (conditional recommendation, low quality of evidence).
12. Biannual screening should be offered to affected infants annually until age 7 years with α-fetoprotein and ultrasounds (conditional recommendation, very low quality of evidence).
Summary of evidence
The phenotype of FAP includes benign and malignant neoplasms in other organs. Other organs commonly affected include the thyroid (with papillary thyroid cancer), adrenal (non functioning adenomas), the small intestine (adenomas or carcinoma), bones (osteomas), retina (congenital hypertrophy of the retinal pigmented epithelium), and skin (epidermoid cysts). However, the most common causes of death in FAP after CRC are duodenal or ampullary cancer and desmoid disease.
Endoscopically visible duodenal adenomas are found in more than half of FAP patients (101, 144, 145). The lifetime risk for duodenal cancer is 3 to 5%, but in some series it has been even higher (81, 101, 127, 144, 146). The age of duodenal cancer diagnosis ranges from 17 to 81 years, with a mean between 45 and 52 years. Approximately half of duodenal cancers are ampullary or periampullary, whereas others are elsewhere in the duodenum (147). Duodenal cancer is one of the leading causes of death in FAP patients who have had prophylactic colectomy (147, 148, 149, 150).
Adenomas beyond the duodenum may occur throughout the small bowel but are concentrated for the most part in the proximal jejunum (50% of cases) and distal ileum (20% of cases) (151, 152, 153). The polyps are most commonly 1 to 10 mm in diameter and multiple. Most duodenal polyps cluster around the ampulla, although in some patients there are small adenomas scattered throughout the duodenum. Adenomas may progress, often slowly, and there is evidence of an adenoma–carcinoma sequence similar to that observed in the colon (101). Adenomas sometimes grow large, exhibit villous histology and increasing degrees of dysplasia, and may cause symptoms. A scoring system has been developed to evaluate the severity of duodenal polyposis and is now widely applied as the Spigelman staging system (154) (Table 9). The risk for duodenal cancer increases to 36% within 10 years for Spigelman stage IV patients (155). The risk of exhibiting Spigelman stage IV duodenal polyposis is 43% by age 60 years and 50% by age 70 years (156). Patients with Spigelman Stage IV duodenal adenomatosis are candidates for a pancreas-preserving duodenectomy. This is much less morbid than a Whipple procedure and patients have a better quality of life. If there is a strong suspicion of cancer, then a Whipple is necessary.

Duodenal adenomatosis staging systema
Gastric fundic gland polyps are also common but gastric adenomas are rare and in western countries gastric cancer is uncommon. Gastric polyps occur in 23 to 100% of FAP patients (101, 126, 144, 157). In the gastric fundus and body, the polyps are most often fundic gland polyps, considered hamartomas. These polyps are histologically seen to consist of simple hyperplasia of the fundic glands with microcysts. Endoscopically, the polyps are multiple sessile lesions, most often 1 to 10 mm in diameter, and are the same color as surrounding mucosa (158). Considerable variation in size and number is observed. The polyps are sometimes so numerous that they coalesce, forming areas of irregular, matted surface mucosa. Fundic gland polyps rarely cause symptoms. Almost half of FAP patients with fundic gland polyps will have superficial dysplasia in some of those polyps (159). Although they are considered nonneoplastic, fundic gland polyps may rarely progress to cancer (160, 161, 162). Adenomatous polyps occur in the stomach of ∼10% of patients with FAP. They are most often confined to the antrum but are occasionally found in the body and fundus.(163, 164) The lifetime risk for gastric cancer in FAP is ∼0.6%, believed both from fundic gland polyps and adenomatous polyps (138).
Upper GI screening has not been demonstrated to improve prognosis but is nonetheless recommended in view of the cancer risk and expectation that mortality can be improved (127, 144, 145, 146, 147, 156, 165). Standard upper endoscopy should be supplemented with a side-viewing instrument to visualize the duodenal papilla. Duodenal screening should begin at age 25–30 years and continue for life, with a frequency determined by the severity of the duodenal polyposis as measured by the Spigelman score. A 0.5–4-year interval for examination is given as follows: (i) every 4 years for Spigelman stage 0; (ii) every 2–3 years for stage I disease; (iii) every 1–3 years for stage II disease; (iv) every 6–12 months for stage III disease; and (v) for stage IV disease: surgical evaluation, expert surveillance every 3–6 months and complete mucosectomy or duodenectomy, or Whipple procedure if duodenal papilla is involved (101, 127). Another approach to screening is every 3-year endoscopy if adenomas are not found and annually if they are. The stomach should be examined during endoscopy and any polyps judged to be of concern because of size, color, or gross appearance biopsied. The role of examination of the small bowel beyond reach of the upper endoscope by computed tomography (CT) enterography, push or balloon enteroscopy, or capsule endoscopy if upper endoscopy demonstrates severe duodenal polyposis is uncertain (153).
In contrast to colorectal polyps and cancer, the expression of upper GI polyps, both gastric and duodenal, does not appear to be attenuated in number, age at emergence, or cancer risk in AFAP compared with FAP (89, 162); therefore, EGD surveillance should also be performed at age 25–30 years, and continued according to the rules stated for classical FAP.
Given that the risk of duodenal cancer in MAP is similar to that of AFAP and FAP, upper GI endoscopy with added side-viewing duodenoscopy should be considered, starting at around age 30 years and repeated at intervals similar to AFAP and FAP, again depending on duodenal findings (166).
Gallbladder, bile ducts, and pancreas
Both adenomatous change and cancer have been reported in the gallbladder, bile ducts, and pancreas (138, 151, 167, 168, 169, 170). Biliary and pancreatic duct obstructions have arisen from both benign and malignant lesions. The cancer risks are shown in Table 7. There are no surveillance strategies that are currently recommended for these malignancies.
Extraintestinal malignancies
Up to 12% of FAP patients have thyroid cancer and 80% have nodular thyroid (171). The mean age of diagnosis of thyroid cancer is 28 years, ranging from 12 to 62 years (172). A female preponderance is observed, and the histology is predominantly papillary, commonly with a cribriform–morular pattern. Annual thyroid ultrasound is recommended for thyroid screening in FAP (171).
Hepatoblastoma occurs in 1.6% of FAP patients, exhibits a male predominance, and associates somewhat with mutations in the 5´ end of the APC gene. This malignancy most often occurs in the first 5 years of life, with some risk up to 15 years of age (173). Screening with every 3- to 6-month serum α-fetoprotein and liver ultrasound for the first 5 to 10 years in FAP patients has been suggested but is still debated. A family history of hepatoblastoma may be an indication to do this from age 6 months to 6 years. A decision to perform hepatoblastoma screening mandates genetic testing in infancy to see if the child carries the mutation.
Desmoid tumors
Screening is not done for desmoids, but evaluation is done for palpable masses and a full work-up for suggestive symptoms. Periodic abdominal imaging is not generally recommended, but preoperative abdominal CT scan before colectomy may be considered if desmoids have been an issue in family members.
The lifetime risk of extracolonic tumors in MAP is not as well defined as the colorectal phenotype. In a large study of 276 MAP patients, 17% had extracolonic lesions, with an estimated 38% lifetime risk of extracolonic malignancy that is approximately double the risk in the general population (174). Similar to FAP and AFAP, the lifetime risk of duodenal cancer in MAP has been estimated to be 4% (174). Although gastric lesions have been found in up to 11% of patients with MAP, data are currently lacking to support an increased risk of gastric cancer (107).
Other cancers such as endometrial, breast, ovarian, bladder, various skin, and thyroid have been reported in patients with MAP (107), although it is still not clear whether the lifetime risk for these malignancies is increased. Although rare, other findings seen in patients with MAP have included sebaceous gland adenomas, carcinomas and epitheliomas, lipomas, congenital hypertrophy of the retinal pigment epithelium, osteomas, desmoid tumors, epidermoid cysts, and pilomatrixomas (107). Surveillance and disease management of the colon in MAP should be similar to patients with multiple adenomas, AFAP, and FAP (166).
In women with a POLD1 mutation, pelvic ultrasound and selective endometrial biopsy may be considered as the POLD1 variant has been associated with endometrial and possibly brain tumors.
Recommendation
13. Postsurgical surveillance should include yearly endoscopy of rectum or ileal pouch, and examination of an ileostomy every 2 years (strong recommendation, low quality level of evidence).
Summary of evidence
Adenomas may develop in the ileal pouch after colectomy with IPAA surgery, or they may develop in the small segment of remaining rectal epithelium after restorative proctocolectomy (175, 176, 177). There appears to be a small but real risk for cancer in the ileal pouch (177, 178, 179). Advanced dysplasia and cancer may occur at the anal transition zone, either from rectal tissue unexpectedly remaining with ileo-anal anastomosis or from the short segment of rectum often remaining with restorative proctocolectomy (143, 150, 180).
After colectomy or proctocolectomy, endoscopic surveillance of the rectum or ileal pouch should continue yearly. Concerning polyposis in the rectum (large adenomas, high-grade dysplasia, >20 adenomas) is treated either by polypectomy or proctectomy. Pouch polyposis can be treated by polypectomy or chemoprevention with sulindac. Ileostomies should be checked every 2 years as adenomas and even cancer can develop on the stoma.
Prevention strategies
Much attention and effort has been given to examining chemoprevention for colonic and duodenal polyps in FAP (127). Considerable regression and prevention of colonic and rectal adenomas has been demonstrated with sulindac, but cancer prevention is less certain. Celecoxib appears to have a more modest effect in the colon and rectum, but some effect in duodenal adenoma regression as well. Celecoxib was approved for use in the United States for several years for FAP, but this indication has now been removed. Concern over cardiovascular side effects of long-term cyclooxygenase-2 (COX-2) inhibitors has dampened enthusiasm for their use in FAP. In view of the uncertainty of cancer prevention with sulindac, it is not considered a substitute for colectomy but has shown utility in rectal surveillance by substantially decreasing the number of adenomas needing removal at periodic examination. Chemoprevention studies examining nonsteroidal anti-inflammatory drugs and other agents continue in the hopes that colectomy might be delayed.
Hamartomatous polyposis syndromes
Peutz–Jeghers syndrome
Indications for genetic testing
Summary statement
- Individuals with perioral or buccal pigmentation and/or two or more histologically characteristic GI hamartomatous polyp(s) or a family history of PJS should be evaluated for PJS.
Summary of evidence
PJS is an autosomal-dominantly inherited syndrome that includes histologically distinctive hamartomatous polyps of the GI tract and characteristic mucocutaneous pigmentation (181, 182, 183, 184). Its incidence is estimated at between 1 in 50,000 and 1 in 200,000 births (185). The mucocutaneous melanin pigment spots are seen in >95% of cases. They are 1 to 5 mm in diameter and most commonly occur in the perioral area and on the buccal mucosa (94%). Pigment spots on the lips are distinctive in that they cross the vermilion border and are often much darker and more densely clustered than common freckles. These spots also occur on the face, forearms, digits, palms, soles, perianal area, and rarely on the intestinal mucosa. The pigment appears in infancy and may fade with age, but less so on the buccal mucosa. GI polyps occur in 88 to 100% of patients. PJS polyps are histologically distinct. They are nondysplastic, have normal overlying epithelium specific to the GI segment in which they are found, and exhibit an arborizing pattern of growth with muscularis mucosae extending into branching fronds of the polyp. Epithelial infolding may result in what is termed pseudoinvasion that can lead to an incorrect diagnosis of cancer. Adenoma and cancer may occur in PJS polyps (186). Their frequency by segment is: stomach, 24%; small bowel, 96%; colon, 27%; and rectum, 24% (183, 186). Polyp sizes range from 0.1 to 3 cm in diameter. Polyp growth begins in the first decade of life, but patients typically do not develop symptoms until the second or third decade (187, 188). Symptoms arise from larger polyps that may infarct, ulcerate, bleed, and cause intestinal obstruction and intussusception, usually in the small intestine.
Genetic etiology
Summary statement
- Genetic evaluation of a patient with possible PJS should include testing for STK11 mutations.
Summary of evidence
PJS arises from mutations of the STK11 gene, a tumor suppressor seronine/threonine kinase gene, previously called LKB1, on chromosome 19p (182, 183). Up to 94% of PJS families have mutations of STK11 with up to a third of disease causing mutations representing large deletions (189, 190). Approximately 25% of newly diagnosed PJS patients represent de novo mutations (183). There do not appear to be genotype–phenotype correlations with mutation location in the STK11 gene (191). Once a disease-causing mutation is identified in a patient with PJS, other family members should undergo mutation-specific testing to determine whether the disease is present or absent so that appropriate surveillance can be undertaken.
Surveillance and management
Recommendation
14. Surveillance in affected or at-risk PJS patients should include monitoring for colon, stomach, small bowel, pancreas, breast, ovary, uterus, cervix, and testes cancers. Risk for lung cancer is increased, but no specific screening has been recommended. It would seem wise to consider annual chest radiograph or chest CT in smokers (conditional recommendation, low quality of evidence).
Summary of evidence
Numerous studies and reviews have now reported a high risk of both GI and extraintestinal cancer in PJS (126, 182, 183, 185, 192, 193, 194, 195, 196, 197). Individual risks by cancer site are given in Tables 5 and 7. The malignant risk in PJS includes colorectal, breast, pancreatic, gynecological, small bowel, lung, and gastroesophageal cancers in that order of risk (198). The overall risk of developing any cancer at ages 20, 30, 40, 50, 60, and 70 years was 1%, 3%, 19%, 32%, 63%, and 81% respectively. In terms of specific cancers, estimated lifetime risks are 39% for colorectal, 29% for gastric, 13% for small bowel 24–54% for breast, 21% for ovary, 10–23% for cervix, 9% for uterus, 9% for testicular, 7–17% for lung, and 11–36% for pancreas (196). Distinctive tumors in women with this condition include ovarian sex cord tumors with annular tubules that are benign, although ∼20% become malignant; mucinous tumors of the ovary; and well-differentiated adenocarcinomas of the uterine cervix, called adenoma malignum (186). Nine percent of males develop large cell calcifying sertoli cell tumors of the testes, resembling sex cord tumors with annular tubules, that have a 10 to 20% chance of becoming malignant (199). Feminization may occur with the benign testicular tumors. Mainly because of cancer, the overall survival of PJS patients is significantly shorter than age- and gender-matched controls (200).
Surveillance guidelines for PJS are empiric and based on the risk for GI complications and cancer. See Table 10 for specific recommendations. A consortium review group has recommended that upper GI endoscopy (EGD) and colonoscopy be done first at age 8 years (182). If polyps are found, both examinations should be repeated every 3 years. If none are found, a second baseline examination should be done at age 18 years and then every 3 years thereafter. Similar surveillance is recommended for the small bowel, i.e., first examine the small bowel by video capsule endoscopy at age 8 years, but then repeat this surveillance every 3 years from that age. Modern CT enterography is accurate at detecting small bowel polyps, particularly those ≥1 cm in diameter, but repeated X-ray exposure is problematic.

Surveillance recommendations for hereditary gastrointestinal (GI) cancer syndromes

Continued.
Treatment involves EGD and colonoscopic removal of polyps (probably all those >0.5 or 1 cm in diameter) (185). Clearing of all polyps is preferable but not always possible. Colectomy is sometimes necessary to control colonic polyps and should be considered if colonoscopic management is difficult and especially if neoplastic change is found in colonic polyps. Intussusception is the primary complication of small bowel polyps, starting at a young age, and continuing throughout life (188). Surveillance and treatment of the small bowel are based in large part on prevention of this complication. In the recent study by van Lier et al. (188), the initial episode of intussusception occurred at a median age of 16 years (range, 3–50 years), with 50% of first episodes presenting by age 20 years. Of all intussusceptions, 80% presented as an acute abdomen and the average polyp size causing this complication was 3.5 cm (range, 15–60 cm). When small bowel intussusceptions occur, surgery is often necessary and should include careful examination of the entire small bowel to eliminate all significant polyps. Intraoperative endoscopy is often a helpful adjunct to accomplish extensive polyp removal. This is also an appropriate time to examine and remove gastric and duodenal polyps of significant size (201). The advent of video capsule endoscopy, double balloon enteroscopy, and CT enterography is changing diagnostic and management approaches to PJS by allowing earlier detection of polyps and nonoperative removal in many cases (202, 203, 204, 205).
Prevention strategies
Chemoprevention approaches to decrease polyp burden in PJS are under study but not yet a reality. PJS polyps exhibit overexpression of COX-2, suggesting that COX-2 inhibitors may be useful in reducing polyps (206). Hyperactivation of the mammalian target of rapamycin has been associated with PJS. In addition, inhibition of mammalian target of rapamycin in a PJS mouse model has demonstrated decreased polyp burden (207). Everolimus, a mammalian target of rapamycin inhibitor, is under study as a potential agent for treatment of PJS (182).
Juvenile polyposis syndrome
Indications for genetic testing
Summary statement
- Individuals with five or more juvenile polyps in the colorectum or any juvenile polyps in other parts of the GI tract should undergo evaluation for JPS.
Summary of evidence
JPS is an autosomal-dominantly inherited condition where multiple juvenile polyps are found in the colorectum (98%), stomach (14%), jejunum and ileum (7%), and duodenum (7%) (183, 208, 209, 210). The incidence of JPS is between 1 in 100,000 and 1 in 160,000 individuals (210). The polyps in JPS vary in size from small sessile nodules to pedunculated lesions that are ≥3 cm in diameter. Most large polyps are pedunculated, but small polyps, especially those in the stomach, are sessile. Grossly, most polyps exhibit a surface that is smooth, rounded, reddish colored, and without fissures or lobulations; large polyps may appear to be multilobulated. A white exudate is often seen on the polyp surface. On cut section, there are cystic spaces filled with mucin. Microscopically, there is abundant lamina propria with benign but often elongated and cystically dilated glands and lack of a smooth muscle core. Excess chronic inflammatory cells are sometimes present. The epithelial lining of the surface and cysts is nondysplastic and reflects the area of the GI tract where the polyp is located. Polyps begin to appear in the first decade of life, and dozens to many hundreds of polyps are present in the fully developed syndrome. Most patients develop symptoms in the first two decades of life. The average age at diagnosis is 18.5 years but may be later. Rectal bleeding with anemia is the most common presenting symptom, followed by abdominal pain, diarrhea, passage of tissue per rectum, and intussusception (183, 208). The majority of colonic polyps, 70% in one study, occurred in the proximal colon (210).
The generally accepted clinical criteria for JPS include: (i) at least five juvenile polyps in the colorectum; (ii) juvenile polyps in other parts of the GI tract; or (iii) any number of juvenile polyps in a person with a known family history of juvenile polyps (210).
Genetic etiology
Summary statement
- Genetic evaluation of a patient with possible JPS should include testing for SMAD4 and BMPR1A mutations.
Summary of evidence
Juvenile polyposis occurs as a result of mutations of the SMAD4 gene (also called the MADH4 gene) or the BMPR1A gene (210, 211, 212, 213, 214). Up to 60% of individuals with clinically defined JPS are now found to exhibit mutations of the SMAD4 or BMPR1A genes (215). Approximately 25% of newly diagnosed cases are sporadic and thus represent new or de novo mutations, whereas 75% will have a family history (183). Fourteen percent of mutations are large deletions and 10% are promoter mutations (215, 216, 217). Both genes are tumor suppressor genes involved in the tumor growth factor-β signaling family and directly or indirectly affect cell growth inhibition and apoptosis. There is evidence that normal bone morphogenetic protein signaling also suppresses Wnt signaling to ensure a balanced control of stem cell self-renewal (218). Biallelic gene inactivation has been noted in both stromal cells and epithelial cells of polyps (208, 219).
Genetic testing is particularly important in JPS, both to confirm the diagnosis in a proband and to test relatives. Testing is also important to separate JPS from other conditions in which juvenile polyps form, especially CS and Bannayan–Riley–Ruvalcaba syndrome. Once a disease-causing mutation is identified in a patient with JPS, other family members should undergo mutation-specific testing to determine whether the disease is present or absent so that appropriate surveillance can be undertaken.
Surveillance and management
Recommendations
15. Surveillance of the GI tract in affected or at-risk JPS patients should include screening for colon, stomach, and small bowel cancers (strong recommendation, very low quality of evidence).
16. Colectomy and IRA or proctocolectomy and IPAA is indicated for polyp-related symptoms, or when the polyps cannot be managed endoscopically (strong recommendation, low quality of evidence).
17. Cardiovascular examination for and evaluation for hereditary hemorrhagic telangiectasia should be considered for SMAD4 mutation carriers (conditional recommendation, very low quality of evidence).
Summary of evidence
JPS mutation carriers have a very high risk for colon cancer and an increased risk for gastric, duodenal, and pancreatic cancers (Tables 5 and 7). The cancer risk in JPS is believed to arise from adenomatous tissue within the juvenile polyp, as up to 50% of juvenile polyps in JPS contain areas of adenomatous change. The risk of colon cancer is 17–22% by age 35 years and approaches 68% by age 60 years (183, 220). The mean age of colon cancer is 34 years, with a range of 15 to 68 years. Gastric cancer risk is 30% in those with SMAD4 mutations (183, 210). The median age of upper GI carcinoma is 58 years, with a range of 21 to 73 years (221, 222).
Surveillance guidelines for JPS are found in Table 10. Colonoscopy should be annual, beginning at age 12 years or earlier if symptoms occur, especially rectal bleeding. It should be repeated every 1 to 3 years depending on polyp burden and polyps ≥5 mm should be removed (210). Upper endoscopy is recommended every 1 to 3 years beginning at age 12 years, or earlier for symptoms, and should be repeated every 1 to 3 years, depending on severity with removal of polyps ≥5 mm. The small bowel past the duodenum should be periodically surveilled, depending on initial polyp findings, by enteroscopy, capsule endoscopy, and/or CT enterography if duodenal polyposis is present or if there is unexplained anemia, protein-losing enteropathy, or other small bowel symptoms. Patients with limited numbers of polyps in any area of the GI tract can usually be managed with endoscopic polypectomy. Colectomy with IRA is indicated if cancer, high-grade dysplasia, or polyposis cannot be adequately controlled endoscopically (183, 208). Surveillance of the remaining rectum or pouch is necessary (223). Proctocolectomy with IPAA may be needed depending on the number of rectal polyps (223). Half of those with IRA will later need proctectomy because of polyp perfusion. Complete or partial gastrectomy may also be necessary for patients with advanced dysplasia, gastric cancer, or even massive gastric polypsis that cannot be effectively controlled endoscopically (183, 208). Other screening should include annual complete blood count, cardiovascular examination, and hereditary hemorrhagic telangiectasia protocol evaluation if SMAD4 mutation is present (210).
Cowden syndrome (PTEN hamartoma tumor syndrome)
Summary statement
- Individuals with multiple GI hamartomas or ganglioneuromas should be evaluated for CS and related conditions.
Summary of evidence
CS and its variants, including Bannayan–Riley–Ruvalcaba syndrome and PTEN hamartoma tumor syndrome (PHTS), have been associated with a broad range of clinical phenotypes. Colonic polyps are found in up to 95% of CS patients undergoing colonoscopy (224, 225). Polyps are few to numerous (even hundreds) and are distributed throughout the colon. The natural history of polyps is not well characterized, although polyps may occur at a young age. Hamartomatous polyps are the most common histologic type, occurring in up to 29% in one study (224). Polyp types include juvenile polyps, ganglioneuromas, adenomas, and inflammatory polyps (224, 226, 227), and less commonly leiomyomas, lipomas, and lymphoid polyps (228). Hyperplastic polyps have also been reported as an association, but have not been observed in all studies (224, 227). The majority of CS patients have multiple synchronous histologic types at colonoscopy.
A frequent finding in the esophagus is diffuse glycogenic acanthosis (228, 229). One or several such lesions may occasionally be observed in patients undergoing EGD for various reasons, but diffuse, sometimes many hundreds of lesions are observed in ≥80% of those with PHTS (227). It has been suggested that diffuse esophageal glycogenic acanthosis combined with colonic polyposis should be considered pathognomonic for CS (228).
Several investigations report the frequent finding of multiple hamartomatous polyps in the stomach, duodenum, and small bowel (183, 224, 229). Similar to the colon, histologies include hamartomas, hyperplastic polyps (different from colonic hyperplastic polyps), ganglioneuromas, adenomas, and inflammatory polyps. An upper GI study of 10 phosphate and tensin homolog (PTEN) mutation-positive patients found all 10 to have multiple hyperplastic gastric polyps and 3 to have multiple hamartomatous polyps in that location (227). One patient had a single hamartomatous polyp in the duodenum whereas three had adenomatous polyps. There are reports of gastric and colon cancers in CS patients (183, 230).
Specific indications for evaluation for CS are delineated in Table 11.

Indications for genetic evaluation for Cowden syndrome (PTEN hamartoma tumor syndrome)a
Genetic etiology
Summary statement
- Genetic evaluation of a patient with possible CS should include testing for PTEN mutations.
Summary of evidence
CS is caused by mutations in the PTEN gene (231, 232, 233, 234). Once a disease-causing mutation is identified in a patient with CS or related conditions, other family members should undergo mutation-specific testing to determine whether the disease is present or absent so that appropriate surveillance can be undertaken.
Surveillance and management
Recommendation
18. Surveillance in affected or at-risk CS patients should include screening for colon, stomach, small bowel, thyroid, breast, uterine, kidney, and skin (melanoma) cancers (conditional recommendation, low quality of evidence).
Summary of evidence
Management of PHTS involves prevention and early detection of the associated cancers through surveillance. Colon cancer has not been associated with CS historically (183), although recent studies have indeed shown increased risk for this malignancy. One multicenter study found 13% of PTEN mutation carriers to have colon cancer, all younger than 50 years of age (224). Investigations have now indicated a 9 to 16% lifetime risk for large bowel cancer (225, 235, 236). It is uncertain whether colon malignancy arises from adenomatous and/or hamartomatous polyps in PHTS, although there is little doubt as to the increased risk and possibility of young age onset.
Surveillance recommendations are provided in Table 10. Recommendations are all expert opinion based rather than evidence based, and derived from screening guidelines of the relevant cancers in other settings but adjusted for the malignancy risks observed in PHTS.
Hereditary mixed polyposis syndrome (HMPS) is a condition that was originally described in a large Ashkenazi Jewish family with multiple colorectal polyps and cancer. Affected patients exhibited mixed juvenile–adenomatous polyps and also adenomatous, hyperplastic, serrated adenomas, and mixed hyperplastic–adenomatous polyps and adenocarcinomas. Mean age of polyp occurrence in one family was 28 years. HMPS may be misdiagnosed as JPS or serrated polyposis syndrome (SPS) and vice versa.
Even though HMPS linked to a locus on chromosome 15q13.3–q14 in a number of families, which includes the CRAC1 gene, the etiology remains elusive. Recently, a duplication 40 kb upstream of the GREM1 gene locus at chromosome 15 was found in two individuals with HMPS. The authors hypothesized that this duplication interacts with the GREM1 promoter causing increased GREM1 expression, resulting in a predisposition to multiple colorectal polyps.
Genetic testing for GREM1 mutation and expression might be considered in families with adenomatous and hamartomatous polyposis in which an etiology cannot be determined. Management from what is now known should probably be similar to that for FAP, depending on the polyp number, size, and histology.
Serrated polyposis syndrome
Clinical definitions
Summary statement
- Individuals who meet at least one of the following criteria have the clinical diagnosis of SPS: (i) at least 5 serrated polyps proximal to the sigmoid colon with ≥2 of these being >10 mm; (ii) any number of serrated polyps proximal to the sigmoid colon in an individual who has a first-degree relative with serrated polyposis; and (iii) >20 serrated polyps of any size, distributed throughout the large intestine.
Summary of evidence
SPS, previously referred to as hyperplastic polyposis syndrome, is a rare condition currently defined by clinical criteria and characterized by a predisposition to serrated polyps and an increased risk of CRC (237, 238, 239). Originally, hyperplastic polyps were the only lesion included in the diagnostic criteria for hyperplastic polyposis (240). In addition to hyperplastic polyps, other serrated polyps including sessile serrated polyps and traditional serrated adenomas may also be found, and hence the preferred term serrated polyposis syndrome (241).
The updated World Health Organization (WHO) diagnostic criteria for SPS include any one of the following: (i) at least five serrated polyps proximal to the sigmoid colon with two or more of them >10 mm in diameter, (ii) any number of serrated polyps proximal to the sigmoid colon in an individual who has a FDR with SPS, or (iii) >20 serrated polyps of any size, but distributed throughout the colon (242). The true prevalence of SPS is not known, but had been previously estimated to be 1:100,000 based on a large screening colonoscopy study of 50,148 participants in which 28 subjects (0.06%) were found to have the syndrome. (243). More recent studies have evaluated the prevalence of SPS based on the WHO criteria. The National Health Service Bowel Cancer Screening Programme (NHSBCSP) reviewed all pathology and colonoscopy records for guaiac fecal occult blood test-positive patients presenting for index screening colonoscopy (244). Out of 755 patients, 5 (0.66%) met SPS criteria 1 and/or 3 (244). Therefore, 1 in 151 patients in the NHSBCSP met SPS criteria during their index colonoscopy, a much higher rate than previous reports of SPS in the general population (244). In a study from Barcelona, the prevalence of SPS in patients undergoing colonoscopy after a positive fecal immunochemical testing was 8 out of 2,355 (0.34%) (245).
Genetic etiology
Summary statement
Indications for genetic testing
- A clear genetic etiology has not yet been defined for SPS, and therefore genetic testing is currently not routinely recommended for SPS patients; testing for MUTYH mutations may be considered for SPS patients with concurrent adenomas and/or a family history of adenomas.
Summary of evidence
Although the genetic etiology of SPS remains unknown, 3 patients were found to meet diagnostic criteria for SPS in a series of 17 biallelic MUTYH mutation carriers (18%) (136). Conversely, only one biallelic MUTYH mutation carrier was observed in a study of 126 patients with SPS (0.8%) (246). In both studies, the patients who met criteria for SPS also reported a history of adenomas. These observations indicate some overlap in the presentation (and potentially the pathogenesis) of MAP and serrated polyposis. Although data are currently limited, it may be reasonable to consider genetic testing for MUTYH mutations in patients with SPS, particularly if adenomas are concurrently seen.
Although the mechanisms are not entirely clear, there seems to be a strong association between smoking and SPS (247). In a small study of 32 SPS patients, the rate of current smoking was 47%, and this was significantly higher than the rate in colonoscopy controls (17%) and population controls (12%) (247). In another study of patients with multiple serrated polyps, many of whom met criteria for SPS, 51 of 88 (58%) were ever smokers (248). It is speculated that smoking, although not the cause of SPS, does intensify the phenotypic expression and therefore may be a modifiable risk factor for colorectal lesions(247).
Various studies have shown that a family history of colorectal and other cancers is common and even increased in patients with SPS. Some have suggested that this supports a hereditary etiology to SPS (249, 250). However, nongenetic causes, referral bias, and chance occurrence should not be overlooked as substantial factors in these studies (251).
Surveillance and management of CRC
Recommendation
19. Patients with serrated polyposis should undergo colonoscopies every 1–3 years with attempted removal of all polyps >5 mm diameter (conditional recommendation, low quality of evidence).
Summary of evidence
It is now well established that SPS is associated with an increased risk for CRC (252). The specific lifetime risk of colorectal in SPS is not well defined as most study cohorts to date are relatively small, phenotypically diverse, and subject to referral bias. It has been estimated that the lifetime CRC risk is >50%, although this is likely an overestimate (243). CRC was diagnosed in 5 of 77 (6.5%) SPS patients after a median follow-up time of 1.3 years (253). Four out of five of these CRCs were found in serrated polyps <20 mm (253). Boparai et al. (253) estimated that the 5-year risk of CRC under surveillance was 7%. In two large descriptive studies of SPS patients, the majority of index cases displayed a pancolonic distribution of polyps (89–96%), the presence of adenomas (78–80%), a diagnosis of CRC (31–42%) and a mean age at diagnosis of 48 years (250, 254). In a smaller prospective series of 13 hyperplastic polyposis syndrome patients, 5 (71%) of the 7 CRCs reported were located in the right colon (237). In another retrospective study of 77 patients with SPS, 22 (28.6%) were diagnosed with CRC at their baseline colonoscopy. Five (6.5%) more patients developed CRC while being followed for hyperplastic polyposis syndrome (253). Synchronous adenomas are found in the majority of patients with SPS (248, 252). Conventional adenomas were more frequently found in patients with CRC than those without (254). In a study of CRCs from SPS patients, 18 of 39 (46%) had the BRAF V600E mutation, 2 of 40 (5.0%) had KRAS mutations, and 17 of 45 (38%) had loss of immunohistochemistry expression of MLH1 and PMS2 (255).
There are no available studies regarding the effectiveness of surveillance in SPS. Based on the reported colon cancer risks, colonoscopy and polypectomy is recommended for individuals who fulfill the WHO definition of SPS. Complete clearance of all polyps ≥1 cm should be done when possible. Subsequent colonoscopy intervals should be determined by the number and size of polyps, as well as the number of concurrent adenomas, but generally should be performed every 1–3 years.
Recommendation
20. Indications for surgery for SPS include an inability to control the growth of serrated polyps, or the development of cancer. Colectomy and IRA is a reasonable option given the risks of metachronous neoplasia (conditional recommendation, low quality of evidence).
Summary of evidence
Surgery is advised when polyps cannot be endoscopically controlled (256). Prophylactic or therapeutic colectomy is indicated with an inability to control the growth of serrated polyps, the presence of high-grade dysplasia in a serrated polyp that cannot be removed in its entirety, or the development of cancer. Subtotal colectomy and IRA is a reasonable option given the risks of metachronous neoplasia.
Surveillance and management of extracolonic malignancies
Recommendation
21. There is no evidence to support extracolonic cancer surveillance for SPS at this time. Screening recommendations for family members are currently unclear pending further data and should be individualized based on results of baseline evaluations in family members (conditional recommendation, very low quality of evidence).
Summary of evidence
The data on extracolonic cancers in SPS are insufficient at this time, although Win et al. (250) reported an increased risk of colorectal and pancreas cancer in relatives of patients with SPS. However, PC was not found in any of the 115 patients with SPS in a study by Kalady et al. (249). In a larger study of 105 patients with SPS and 341 FDRs, no increased risk of extracolonic malignancies were seen in patients or their relatives (257). Hazewinkel et al. (257) concluded that extracolonic cancer in SPS and their FDRs is not increased compared with the general population.
Familial cases of SPS have been reported, although infrequently in most studies. However, a recent prospective study of 78 FDRs of patients with SPS found that the incidence of SPS was 32% (258). Only one of these relatives was diagnosed with CRC during screening colonoscopy (258). Other studies have reported a family history of CRCs in 50–59% of FDRs of patients with SPS (241, 248). In one of these studies, only 2 (5%) of the 38 probands with SPS reported a family history of SPS (241). In a recent study, Boparai et al. (259) FDRs of serrated polyposis patients had five times the incidence of CRC, suggestive of a hereditary disorder.
Surveillance recommendations for individuals with a family history of serrated polyposis are currently unclear, pending further data. It is reasonable to screen FDRs at the youngest age of onset of serrated polyposis diagnosis (after the exclusion of other genetic causes), and subsequently per colonoscopic findings (248). The frequency and age of initiation of colonoscopic screening in at-risk family members of patients with SPS is less clear. Oquinena et al. (258) recommended that colonoscopy in FDRs start at age 35 years (or 10 years before the youngest age of SPS onset in the family, whichever comes first) and then every 3–5 years if no polyps are found. If the FDRs are found to meet SPS criteria 1 or 3, annual surveillance should be performed or every 2 years if only SPS criteria 2 is met (258). On the contrary, the National Comprehensive Cancer Network (NCCN) recommends that FDRs should have colonoscopy at the earliest of the following: (i) age 40 years, (ii) same age as the youngest SPS diagnosis in the family, and (iii) 10 years before CRC in the family in a patient with SPS (260). Further work is ongoing to better define the cancer risks in probands and their relatives so that accurate risk stratification and risk recommendations can be made regarding SPS.
Prevention strategies
Summary of evidence
Observational studies suggest a strong association between smoking and SPS. Independent studies have reported significant smoking history in patients with SPS, leading to the assumption that smoking may be a modifiable risk factor in the pathogenic pathway of colorectal lesions (248, 250).
Hereditary pancreatic cancer
Clinical definitions
Summary statement
- Individuals should be considered to be at risk for familial pancreatic adenocarcinoma if they: (i) have a known genetic syndrome associated with pancreatic cancer, including hereditary breast–ovarian cancer syndrome, familial atypical multiple melanoma and mole syndrome (FAMMM), PJS, LS, or other gene mutations associated with an increased risk of pancreatic adenocarcinoma; (ii) have two relatives with pancreatic adenocarcinoma, where one is a FDR; (iii) have three or more relatives with pancreatic cancer; or (iv) have a history of hereditary pancreatitis.
Summary of evidence
The known hereditary syndromes associated with an increased risk for developing PC along with their relative risk for developing PC are shown in Table 12. PC is one of the key cancers used in determining whether a patient’s family history warrants genetic risk evaluation for hereditary breast and ovarian cancer syndrome according to the NCCN Guidelines for Genetic/Familial High-Risk Assessment: Breast and Ovarian (3). Referral for genetic assessment for FAMMM syndrome should be considered in a PC patient with a personal or family history of melanoma or multiple dysplastic nevi (261). Patients who meet the aforementioned criteria for LS, PJS, or FAP may also be at increased risk for PC. Consideration for genetic counseling for testing for hereditary pancreatitis is based on expert opinion and warranted for PC patients with a personal history of at least 2 attacks of acute pancreatitis of unknown etiology, a family history of pancreatitis, or early-age onset chronic pancreatitis (262, 263).

Syndromes associated with pancreatic adenocarcinoma and risk of developing pancreatic cancer
The risk for development of PC in those patients with an inherited predisposition for PC development is shown in Table 12. The risk is highest in hereditary pancreatitis (53-fold risk) (264), PJS (132-fold risk) (197), and FAMMM families with a known CDKN2A mutation (13- to 39-fold risk) (265, 266, 267). The risk is not as great for BRCA1 (˜2-fold) (268), BRCA2 (3- to 9-fold) (269, 270), LS (9- to 11-fold) (71, 271), and ATM (˜3-fold) (272) mutation carriers based on registry data. There are currently no data available on the risk of developing PC in PALB2 mutation carriers.
Familial pancreatic cancer (FPC) has been defined by consensus opinion as families with ≥2 FDRs relatives who do not meet criteria for a known PC-associated hereditary syndrome (262). Case reports first demonstrated families with an excess number of PC cases. Best estimates from case–control and cohort studies suggest that up to 10% of PC patients will have a first- or second-degree relative with PC (273, 274). The risk for developing PC in FPC kindreds is dependent on the number of FDRs as shown in Table 12. Segregation analysis of families with multiple PC cases has shown an autosomal-dominant inheritance pattern and a 32% lifetime risk for PC development at age 85 years (275). The prospective risk for PC development in individuals with FPC is related to the number of FDRs with PC in the kindred. Those with one or two PC-affected FDRs had a risk ranging from 4- to 7-fold, although whereas those with ≥3 PC-affected FDRs had a risk ranging from 17 to 32-fold (262, 276, 277). The complexity in cancer risk assessment has led to the development of a risk prediction model (PancPRO) to provide more detailed risk estimates for individuals from FPC kindreds that take into account the ages at diagnosis, family size, and the relationship between family members (278).
Genetic etiology
Summary statement
- Genetic testing of patients with suspected FPC should include analysis of BRCA1/2, CDKN2A, PALB2, and ATM. Evaluation for PJS, LS, and hereditary pancreatitis-associated genes should be considered if other component personal and/or family history criteria are met for the syndrome.
Summary of evidence
In attempts to discover the cause of their PC susceptibility, studies have been performed on familial FPC kindreds for known candidate genes that fall into two groups: (i) genes that cause inherited disorders that are associated with increased risk of PC development (e.g., BRCA1, BRCA2, and CDKN2A) even in the absence of meeting criteria for these hereditary syndromes (279, 280, 281) and (ii) recently described genes such as palladin (PALLD) (282), ATM (283), and PALB2 (284, 285) that were discovered by whole genome sequencing or linkage analysis of FPC kindred(s). As shown in Table 13, results vary depending on the study population with mutations, for example, in BRCA1 ranging from 0 to 6% (281, 286), BRCA2 ranging from 0 to 6% (281, 287, 288), CDKN2A ranging from 0 to 20% (280, 281, 288), and PALB2 ranging from 0 to 5% (281, 285, 288). PALLD was found to be the susceptibility mutation in a large well-characterized family at the University of Washington (282); however, subsequent studies have not found this gene to be responsible for other FPC families (289, 290). Results from a recent study found ATM mutations in 2.4% of FPC families (283). Known genetic mutations are responsible for ˜20% of the familial clustering of pancreatic cancer. Thus, in the majority of cases, the responsible inherited factor(s) accounting for the increased number of PC cases in these kindreds have not been identified.

Yield for germline mutations in familial pancreatic cancer (FPC) kindreds without criteria for known genetic syndromesa
Surveillance and management
Recommendations
22. Surveillance of individuals with a genetic predisposition for pancreatic adenocarcinoma should ideally be performed in experienced centers utilizing a multidisciplinary approach and under research conditions. These individuals should be known mutation carriers from hereditary syndromes associated with increased risk of PC (Peutz–Jeghers, hereditary pancreatitis, FAMMM) or members of FPC kindreds with a PC-affected FDR. Because of a lower relative risk for pancreatic adenocarcinoma development in BRCA1, BRCA2, PALB2, ATM, and LS families, surveillance should be limited to mutation carriers with a first- or second-degree relative affected with PC (conditional recommendation; very low quality of evidence).
23. Surveillance for PC should be with endoscopic ultrasound and/or MRI of the pancreas annually starting at age 50 years, or 10 years younger than the earliest age of PC in the family. Patients with PJS should start surveillance at age 35 years (conditional recommendation, very low quality of evidence).
Summary of evidence
It is not feasible to screen for PC because of its low incidence in the general population with an estimated lifetime risk in the United States of 1.4% in 2013 (291). Expert opinion has recommended that individuals with a relative risk of more than fivefold when compared with the general population warrant consideration for PC surveillance (73, 262, 292), with the aim to detect early pancreatic lesions that can be intervened upon. Two such precursor lesions of PC include intraductal papillary mucinous neoplasms and pancreatic intraepithelial neoplasia (293).
Based on this degree of risk, candidates for PC surveillance are limited at this time to unaffected individuals from pancreatic cancer-prone families who are candidates for pancreatic surgery. Unlike colon cancer, in which colonoscopy has been proven to be an effective screening tool to reduce colon cancer-related mortality, there is no proven strategy for PC (294). As stated in a recent International Cancer of the Pancreas Screening Consortium summit (73), recommendations for screening patients with a family history of PC “are primarily based on evidence of increased risk, rather than a proven efficacy of screening.” In light of these acknowledged limitations in PC screening, expert opinion has repeatedly emphasized the importance of performing PC surveillance in the setting of active peer-reviewed research protocols by experienced centers utilizing a multidisciplinary team approach (73, 262, 292).
Attempts at a unified recommendation for age to commence screening have been unsuccessful with no clear consensus achieved despite being addressed at two international meetings (73, 262). Two recently reported surveillance studies have shown that the majority of significant lesions have been found in older patients (295, 296). Canto et al. (295) reported in their large multicenter surveillance study of high-risk individuals that the prevalence of pancreatic lesions was age related with a significant difference found in the number of lesions detected between patients <50 years old when compared with patients ≥50 years old. Furthermore, all pancreatic lesions with high-grade dysplasia were in patients >65 years of age. Ludwig et al. (296) also reported differences in finding a significant abnormality based on age, with a yield of 35% in those >65 years old as compared with only 3% in those ≤65 years old. Several studies in both the hereditary and sporadic setting have found that smokers have an earlier age of PC diagnosis as compared with nonsmokers (297, 298, 299), but no data exist regarding whether smoking status should affect surveillance strategy.
Endoscopic ultrasound and MRI/magnetic resonance cholangiopancreatography are the preferred imaging modalities for surveillance as they do not involve the use of radiation like CT scanning and are significantly more accurate in finding pancreatic lesions, particularly small cystic lesions, based on data from CAPS3 study (295).
Recommendation
24. Because of the increased risk for PC development when compared with a pancreatic cyst in the sporadic setting, cystic lesion(s) of the pancreas detected during surveillance of a hereditary pancreatic cancer-prone family member requires evaluation by centers experienced in the care of these high-risk individuals. Determining when surgery is required for pancreatic lesions is difficult and is best individualized after multidisciplinary assessment (conditional recommendation, low quality of evidence).
Summary of evidence
The most common findings in surveillance studies are cystic lesions in the pancreas (295, 300). Management of these cysts is unclear as similar to cysts in a nonhereditary setting, most are benign or just have low-grade dysplasia (73, 292). Recent consensus recommendations propose that these patients be followed according to international consensus guidelines for sporadic branch duct intraductal papillary mucinous neoplasms (301), although the majority agreed that surgery should be considered for those branch duct intraductal papillary mucinous neoplasms ≥2 cm in size (73).
Prevention strategies. As smoking has been shown to be an independent risk factor for PC in families with FPC, all high-risk individuals should be counseled against smoking (302). In individuals with hereditary pancreatitis, in addition to smoking cessation, a low-fat diet should also be recommended.
Hereditary gastric cancer
Hereditary diffuse gastric cancer
Clinical definitions
Summary statement
- Individuals with (i) ≥2 cases of diffuse gastric cancer, with at least one diagnosed at <50 years, (ii) ≥3 cases of documented diffuse cancer in first- or second-degree relatives independent of age of onset; (iii) diffuse gastric cancer diagnosed at <40 years; and (iv) a personal or family history of diffuse gastric cancer and lobular breast cancer with one diagnosed at <50 years should be evaluated for hereditary diffuse gastric cancer.
Summary of evidence
It is estimated that there were ∼21,600 new cases of gastric cancer in the United States in 2013 (303). The lifetime risk for gastric cancer is ∼0.9% (303). The majority of gastric cancer cases are sporadic, but familial clustering is present in ∼10% of cases. There are two main types of gastric cancer: diffuse and intestinal. Intestinal-type gastric cancer is a component tumor in LS, familial adenomatous polyposis, and PJS. These syndromes have already been described previously in this guideline, including the risks for gastric cancer, surveillance guidelines for gastric cancer, and genetic testing recommendations, and therefore intestinal forms of gastric cancer will not be discussed in this section.
The only hereditary cancer susceptibility syndrome known to cause diffuse gastric cancer is hereditary diffuse gastric cancer (HDGC). This condition was originally described in three Maori families with autosomal-dominant diffuse gastric cancer in New Zealand in 1964 (304). Approximately 1–3% of diffuse gastric cancers are attributable to HDGC. A genetic evaluation for HDGC is indicated for families having individuals with (i) ≥2 cases of diffuse gastric cancer, with at least one diagnosed at <50 years, (ii) ≥3 cases of documented diffuse cancer in first- or second-degree relatives independent of age of onset; (iii) diffuse gastric cancer diagnosed at <40 years; and (iv) a personal or family history of diffuse gastric cancer and lobular breast cancer with one diagnosed at <50 years (305).
Genetic etiology
Summary statement
- Genetic testing of individuals who fulfill HDGC clinical criteria should include analysis of CDH1 mutations.
Summary of the evidence
Mutations in the E-cadherin gene (CDH1) were found to be the cause of HDGC in 1998 through linkage analysis (306). The CDH1 gene encodes for the cell-to-cell adhesion protein E-cadherin. This initial finding led to subsequent studies confirming the presence of CDH1 mutations in other gastric cancer families (307, 308, 309). Genetic testing for the CDH1 gene is indicated for individuals from families who meet HDGC criteria and should be initiated in an affected individual if at all possible. CDH1 testing should include sequencing analysis (3–50% of mutations are sequence changes) and deletion/duplication analysis (4% of mutations are large rearrangements) (305). Between 10.5 and 47% of individuals meeting these criteria will be found to have a CDH1 mutation (305). Once a mutation has been identified in a family, all at-risk individuals should be tested beginning at age 16 years, given the early ages of gastric cancer diagnosis in some families (310).
Surveillance and management
Recommendation
25. Management for patients with HDGC should include (i) prophylactic gastrectomy after age 20 years (>80% risk by age 80 years); (ii) breast cancer surveillance in women beginning at age 35 years with annual mammography and breast MRI and clinical breast examination every 6 months, and (iii) colonoscopy beginning at age 40 years for families that include colon cancer (conditional recommendation, low quality of evidence).
Summary of the evidence
An international consortium study of 11 families with at least 3 cases of diffuse gastric cancer who had tested positive for a CDH1 gene mutation found that the lifetime risk of developing gastric cancer was 67% for males and 83% for females, with a mean age at diagnosis of 38–40 years of age (range, 14–85) (311). Females also had an increased risk of lobular breast cancer with a lifetime risk of 39% (311). A recent analysis by an international consortium suggests that the risk of gastric cancer is 80% for both men and women, and that the risk of lobular breast cancer is 60% in women (312). There is some evidence that individuals with HDGC are also at increased risk for signet ring cell colon cancer, although exact risk estimates are not known (313). There is a high rate of gastric cancer detection at the time of prophylactic gastrectomy. A recent systematic review (305) found that of 220 previously reported patients who tested positive for CDH1 mutations, 76.8% underwent a prophylactic gastrectomy whereas 23.2% declined to undergo prophylactic surgery. Among the 169 patients who underwent surgery, 62.7% had negative preoperative endoscopic biopsies and 12.4% (21) tested positive for cancer in their preoperative screening. No information was available on 42 patients. Following gastrectomy, 87% (147) patients had positive histopathology results (including early lesions such as foci of signet ring cells to advanced lesions such as linitis plastica). Only 10% (17 patients) had negative final pathology and it was not reported in 5 patients. It is important to identify both the esophageal and duodenal mucosa at the ends of the surgical specimen because there has been a report of gastric cancer after prophylactic gastrectomy (314).
Gastric cancer surveillance may be used before prophylactic gastrectomy and for patients who decline gastrectomy, but the efficacy is uncertain and should be used with caution. Given the penetrance of this condition, it is recommended that these individuals undergo screening every 6–12 months beginning 5–10 years before the earliest cancer diagnosis in the family (313, 314). It is recommended that this is a detailed 30-min endoscopic examination of the gastric mucosa with multiple random biopsies (313, 314). Some studies have shown an improved detection rate of early gastric cancer with indigo-carmine staining or a pH-sensitive congo red dye followed by pentagastrin stimulation (315)
Because of limited data, the breast cancer surveillance guidelines are based on those for women with a BRCA1 or BRCA2 mutation. Given that lobular breast cancers can be difficult to diagnose by clinical examination and mammography, annual MRI is recommended as part of the surveillance regimen. The colonoscopy guidelines are also based on limited evidence at present.
Informed consent
Genetic testing should only be done in the setting of pre- and posttest genetic counseling. Full informed consent should always be part of the process of cancer genetic counseling. Nongenetics professionals should consider offering cancer genetic testing only if they are able to provide or make available adequate genetic education and counseling as well as access to preventive and surveillance options. Otherwise they should consider referring the patient and family for these services. Standards for informed consent that should be adopted in gastroenterology practices are outlined in Table 14.

Emergence of new genetic testing technologies
The advent and commercial availability of next-generation sequencing panels have increased both the complexity and the ease of cancer genetic testing significantly in the past few years. Next-generation sequencing allows for analysis of multiple genes at one time for a lower cost than traditional Sanger sequencing. There are multiple cancer gene panels including anywhere from 6 to 52 genes now offered at a variety of diagnostic laboratories. Some limitations of the new technology include the longer turnaround time (results can take up to 3 months instead of 2–3 weeks) and a higher chance of finding uncertain results (known as variants of uncertain significance) given the large number of genes included. In addition, there are some genes on these panels for which there are very little data available about the associated cancer risks or appropriate management and others for which the cancer risks are so low that the family would be managed based on the cancer history and not the mutation result. In these cases, the results are not useful clinically and can cause confusion for the patient. However, gene panels also may streamline testing for individuals at increased risk for a cancer susceptibility syndrome and reduce greatly the need for sequential tests where multiple diagnoses are under consideration. In general, genetics professionals are using the next-generation sequencing panels for patients with a long list of differential diagnoses (e.g., if testing for >1 possible genetic syndrome) because the panels are much more cost effective and can shorten the diagnostic journey. It is conceivable that gene panels may in a short period of time replace in large part individual genetic tests and that testing for all the syndromes discussed in this guideline will soon be done simultaneously for at-risk patients.
CONCLUSION
Several well-established hereditary cancer syndromes now exist, each with implications for specific cancer risks in GI and other organ systems. The assessment for cancer susceptibility should be a standard part of patient evaluations in gastroenterology office and endoscopy practices. A systematic and focused family history of cancer and premalignant conditions is the first step and sufficient to screen for ∼10–15% of patients who may need more detailed risk assessment by more extensive family history assessment, genetic counseling, and genetic testing. The underlying genetic etiologies for several syndromes are now well established, and the array of cancer susceptibility genes is continually expanding. Genetic testing is widely available and should be part of standard of care of patients at increased risk for a hereditary cancer syndrome. Mutation carriers and at-risk individuals require intensive surveillance, possibly prophylactic surgery, and family counseling, and management needs to be individualized based on the syndrome under consideration, as well as the specifics of the family history at hand. There is a dire need of organized collaborative international efforts to study benefits of surveillance and surgical strategies in patients with these relatively rare syndromes in order to be able to offer truly evidence-based management recommendations.
ACKNOWLEDGMENTS
This guideline was produced in collaboration with the Practice Parameters Committee of the American College of Gastroenterology. The Committee gives special thanks to Christine Y. Hachem, MD, FACG, who served as guideline monitor for this document. We are grateful to Christine Hachem, Lauren Gerson, and Maria Susano for assistance with guideline development, to Paul Moayyedi for reviewing and grading recommendations, and to Chinedu Ukaegbu for help with drafting of the manuscript tables and assembling reference libraries.
REFERENCES