ACG Clinical Guideline: Evaluation of Abnormal Liver Chemist… : Official journal of the American College of Gastroenterology

INTRODUCTION
The authors were invited by the Board of Trustees and Practice Guidelines Committee of the American College of Gastroenterology to develop a practice guideline regarding the evaluation of abnormal liver chemistries. We used the following resources:
- A formal review and literature search of the world literature on MEDLINE and EMBASE databases dealing with the evaluation of abnormal liver chemistries, studies that dealt with normal or reference range for alanine aminotransferase (ALT) levels and what thresholds trigger an evaluation for actionable liver disease. Studies detailing the relationship between ALT and nonalcoholic fatty liver disease, as well as studies assessing the significance of elevated liver chemistries on overall mortality and morbidity.
- Guideline policies of the American College of Gastroenterology.
- The experience of the authors and independent reviewers, as well as communication with senior hepatologists across the United States with regard to the threshold for evaluating abnormal liver chemistries.
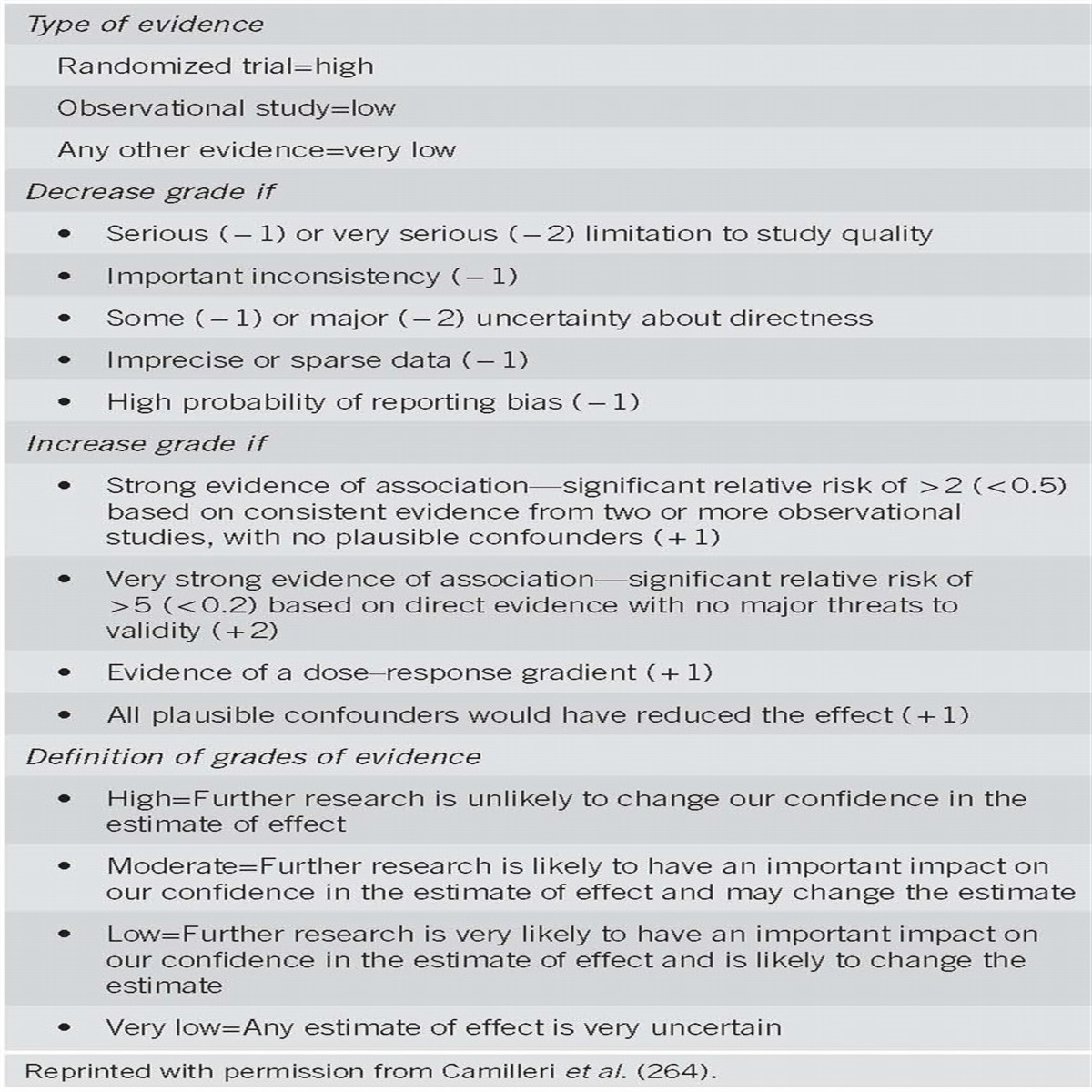
These recommendations are intended for use by physicians and health care providers and suggest preferred approaches to the diagnoses and evaluation of those with abnormal liver tests (Table 1). These guidelines are intended to be flexible and should be adjusted as deemed appropriate when applied to individual patients. Recommendations are evidence-based where possible. On subjects lacking rigid scientific data, recommendations are made based on the consensus opinion of the authors. To more fully characterize the available evidence reporting the recommendations, the ACG Practice Guideline Committee has adopted the classification used by the grading of recommendation assessment, development, and evaluation workup with modifications. The strength of recommendations are classified as strong or conditional. The quality of evidence supporting strong or weak recommendations are designated by the following level is high, moderate low, or very low quality (1). This is a practice guideline rather than a review article.

Recommendations
Liver chemistries that are commonly ordered in comprehensive metabolic profiles are indirect markers of hepatobiliary disease. They are not true measures of hepatic function and thus are best referred to as liver chemistries or liver tests, and should not be referred to as liver function tests. True tests of liver function are not commonly performed but include measurement of hepatic substrates that are cleared by hepatic uptake, metabolism, or both processes (2). Because of the widespread use of the comprehensive metabolic profile testing that is done in routine practice to screen those who present for routine evaluation as well as those who are symptomatic and/or referred for elevation of abnormal liver chemistries, such abnormalities require a rational approach to interpretation. To date, there are no controlled trials that have been performed to determine the optimal approach to evaluate abnormal liver chemistries. This guideline has been developed to assist gastroenterologists and primary care providers in the interpretation of normal and abnormal liver chemistries as well as an approach to prioritize and evaluate those who present with abnormal liver chemistries.
Summary statements:
- Liver chemistries including ALT, aspartate aminotransferase (AST), alkaline phosphatase and bilirubin are markers of liver injury, not liver function, and should be referred to as liver chemistries, or liver tests.
- Albumin, bilirubin, and prothrombin time are markers of hepatocellular function that can be influenced by extrahepatic factors.
- The laboratory measurements of ALT, AST, and alkaline phosphatase are highly reproducible.
- Elevations of AST and/or ALT, alkaline phosphatase, and bilirubin suggest hepatocellular injury and are the abnormal liver chemistries that require assessment and potential evaluation.
- ALT is a more specific marker of hepatic injury than AST.
- An elevated alkaline phosphatase level of hepatic origin may be confirmed by elevation of gamma-glutamyl transferase (GGT) or fractionation of alkaline phosphatase.
The standard comprehensive metabolic profile panel includes AST, ALT, alkaline phosphatase, bilirubin, and albumin. In addition, a prothrombin time may be ordered. Aminotransferases including AST and ALT are enzymes involved in the transfer of amino groups of aspartate and alanine to ketoglutaric acid and are markers of hepatocellular injury and are also referred to as transaminases (3). AST is present in the liver and other organs including cardiac muscle, skeletal muscle, kidney, and brain. ALT is present primarily in the liver, and thus is a more specific marker of hepatocellular cell injury (4, 5, 6). AST increase without elevation in ALT is suggestive of cardiac or muscle disease.
Alkaline phosphatase is part of a family of zinc metalloproteinases enzymes that catalyze the hydrolysis of phosphate esters at an alkaline pH (7). This enzyme is found in hepatocytes on the canalicular membrane, not the bile duct cell. In addition to being present on the canalicular membrane of the hepatocyte, alkaline phosphatase is also found in bone, placenta, intestine, and kidney with the most common extrahepatic location originating from bone. Although rarely used in practice, in those with blood type O and B, serum alkaline phosphatase may increase after a fatty meal due to increased levels of intestinal alkaline phosphatase (8). Alkaline phosphatase may be elevated during pregnancy due to placental synthesis of alkaline phosphatase. Typically, alkaline phosphatase elevates with obstruction of the bile ducts, which is due to increased canalicular synthesis of alkaline phosphatase with subsequent translocation to the sinusoid and is also a measure of liver injury (9). This occurs even if the obstruction is minor and insufficient to increase serum bilirubin levels. To confirm hepatic origin of alkaline phosphatase, the canalicular enzyme GGT may be measured. An elevated GGT suggests that the alkaline phosphatase elevation is of hepatic origin (6). Alkaline phosphatase may also be fractionated to better delineate bone, intestinal or hepatic origin of an elevated alkaline phosphatase. In children and the elderly, alkaline phosphatase levels increase, especially females over 50 years of age, in part due to bone turnover (10, 11).
Bilirubin comes from the breakdown of senescent red blood cells and predominantly circulates in its unconjugated form tightly bound to albumin. Unconjugated bilirubin is not excreted in the urine. Conjugation by uridine 5’-diphospho (UDP)-glucuronosyltransferase makes bilirubin water-soluble (conjugated bilirubin), allowing it to be excreted in bile where it is converted by bacteria in the colon to urobilinogen, which is subsequently excreted in the urine and stool. The absence of urobilinogen gives stool its classic clay-colored appearance in those with impaired bile flow. Unconjugated bilirubin is reported as indirect bilirubin as determined by the van den Bergh reaction and accounts for ˜70% of the total serum bilirubin (12). The total serum bilirubin is usually <1.1 mg/dl and an elevated direct bilirubin (conjugated bilirubin) indicates hepatocellular dysfunction or cholestasis. Fractionation of the bilirubin level to conjugated and unconjugated forms is not done routinely as many laboratories only report total serum bilirubin, which is the sum of conjugated and unconjugated portions. Fractionation of total bilirubin is most helpful when the ALT, AST, and alkaline phosphatase levels are normal or near normal. If the total bilirubin is elevated and fractionation shows the majority of the elevation is unconjugated bilirubin, hepatocellular disease is unlikely to be the explanation. Conjugated bilirubin elevations are present in hepatocellular disorders as well as cholestatic disorders with impairment in bile flow. The delta bilirubin is derived from the reaction of conjugated bilirubin and albumin and has a half-life similar to albumin (5). The delta bilirubin accounts for the prolonged jaundice noted in patients recovering from hepatitis or significant obstruction as its decay is directly related to the half-life of albumin which is 3 weeks.
Two markers of hepatocellular function are albumin and prothrombin time. Albumin is a plasma protein exclusively synthesized by the liver with a circulating half-life of 3 weeks (6). A reduction in albumin (normal ≥3.5 g/dl) usually indicates liver disease of more than 3 weeks duration, although any significant illness can decrease albumin levels due to cytokine effects. Prothrombin time is a far more sensitive measure of liver function than albumin because prothrombin time may be prolonged in patients with severe liver disease of <24 h duration (6). Prothrombin time measures the extrinsic pathway of coagulation. The prothrombin time is a measurement of the clotting tendency of the blood and measures factors 1, 2, 5, 7, 9, and 10. Because factors 2, 7, 9, and 10 are vitamin K dependent, the presence of cholestasis, where vitamin K is not absorbed, will prolong the prothrombin time. Also, significant hepatocellular dysfunction can result in prolongation of the prothrombin time. This does not typically occur until concentrations of clotting factors fall below 10% of normal. As a general rule, in the absence of liver disease, a prothrombin time that is prolonged is due to vitamin K deficiency and/or steatorrhea. It should be noted that prothrombin time can also be elevated with warfarin, heparin bolus, disseminated intravascular coagulation (DIC), and hypothermia.
This practice guideline will discuss the interpretation and evaluation of those with elevation of the major chemistries including ALT, AST, alkaline phosphatase, and bilirubin. Other liver tests (including GGT, albumin and prothrombin time) will be incorporated into the evaluation of these major liver chemistries but will not be discussed separately.
What are truly normal liver chemistry tests?
Summary statements:
- A true healthy normal ALT level in prospectively studied populations without identifiable risk factors for liver disease ranges from 29 to 33 IU/l for males and 19 to 25 IU/l for females, and levels above this should be assessed by physicians.
- Elevated ALT or AST above the upper limit of normal (ULN) in a population without identifiable risk factors is associated with increased liver-related mortality.
- There is a linear relationship between ALT level and body mass index (BMI) that should be assessed by physicians.
- A normal ALT level may not exclude significant liver disease.
- ALT levels are higher in males than females.
- AST and ALT ULN ranges can vary between different labs.
- Clinicians may rely on local lab ULN ranges for alkaline phosphatase and bilirubin.
Normal lab values are generally defined as the mean value of a healthy population±2 s.d.’s. This incorporates 95% of subjects. By definition, 2.5% of the population will be greater than the ULN of the reference population. For alkaline phosphatase and bilirubin levels, establishing normal liver enzyme levels that can be replicated across different reference labs has not been reported as problematic, which differs from the wide variations in ranges reported as normal for ALT levels. However, establishing normal ranges for ALT and AST levels have been problematic due to differences in the definition of healthy control populations that are used to establish the normal reference ranges. One report examined the local reference laboratory ranges for ALT used by the non-alcoholic steatohepatitis (NASH) Clinical Research Network and demonstrated significant differences in the defined ALT ULN (range 35–79 IU/l for men and 31–55 IU/l for women) (13). These wide ranges appeared to be due to the use of different reference populations utilized by the different laboratories with local populations used to establish the normal range of ALT, apparently without consideration of factors such as BMI. Another study found that 67 reference laboratories within one state used different ALT ULN levels ranging from 31 to 72 U/l (ref. 14). In this report, the majority of the labs used equipment from one of four manufacturers, but used different methods to define the ULN, with 40% utilizing manufacturer’s recommendations and local healthy control testing, 38.5% only using the manufacturer’s recommendations, 17% only using local healthy controls, and 8% using published normal levels from textbooks. However, inter-laboratory differences for ALT levels have not been reported to differ significantly (13, 15). When defining a normal population to be used for the establishment of a reference range, the possible presence of underlying liver disease must be considered. Conditions such as non-alcoholic fatty liver disease (NAFLD), viral hepatitis, alcoholic liver disease and the use of medications and herbal agents or supplements need to be factored into the development of these normal ranges. Most importantly, multiple studies have demonstrated that ALT levels correlate with increasing BMI (16, 17, 18).
Determining an ALT level that is normal is clinically relevant to practicing clinicians and patients as there is substantial clinical significance to these different reference ranges for ALT levels between labs. Both practice guidelines and diagnostic and therapeutic studies base clinical decisions including evaluation of abnormal liver tests, therapy for hepatitis B, and evaluation of potential drug-induced liver injury on multiples of the ULN of ALT (19, 20, 21, 22, 23).
Several studies and guidelines have proposed a standardized ULN for ALT based on prospectively acquired data using various methodologies (Table 2). These studies defined normal reference populations by excluding subjects with viral hepatitis, high-risk behavior and NAFLD risk factors (elevated BMI, triglycerides, glucose, and cholesterol). These proposed normal ALT values are lower than commonly reported reference ranges and differ by gender. Of the major liver chemistries, there is sufficient data on the measurement of ALT levels. The true ULN for ALT was proposed in a large study of 6,835 blood donors with normal viral serologies, and BMI under 24.9 kg/m2 to be 30 IU/l for men, and 19 IU/l for women (24). In a Korean study of 1,105 potential liver donors with normal liver biopsies, they reported that age, BMI, and metabolic factors significantly affected ALT levels (25). They proposed ULN for ALT to be 33 IU/l for men and 25 IU/l for women. In an examination of the National Health and Nutrition Examination Survey (NHANES) 1999–2002 and 2005–2008 databases, after eliminating subjects with viral hepatitis, significant alcohol use, diabetes, BMI>25, or enlarged waist circumference, and using statistical analysis, the calculated “maximum correct classification” for ULN of ALT was found to be 29 IU/l for men and 22 IU/l for women (26).

Summary of studies proposing ULN for ALT and or AST levels
Elevated aminotransferase levels and the effect on morbidity and mortality
There is an accumulating set of data demonstrating that AST and ALT elevations correlate with morbidity and mortality (Table 3). An initial report from Germany noted that those with AST>18 U/l had a 3X increased risk of all-cause mortality (27). A Korean study found that, compared with men with AST or ALT<20 IU/l, the 30–39 IU/l group had an 8X (AST) or 9.5X (ALT) relative risk (RR) for liver-related death (28). Similar results were demonstrated from a study comparing the standardized mortality ratios in subjects from Olmsted County where higher ALT levels correlated with higher mortality with the standardized mortality ratio being 0.95 for normal ALT (defined as ULN 45 IU/l for men, 29 IU/l for females), 1.32 for 1–2X ULN, and 1.78 for >2X ULN with a similar relationship for AST levels (29).

ALT and AST levels and liver related mortality
Studies have used the data from the NHANES databases to assess risk of morbidity and mortality in relationship to abnormal liver tests with one study demonstrating that elevated ALT (ULN defined as 30 U/l for men and 19 U/l for women) was associated with significant increases in liver-related mortality (11.2X) and diabetes-related mortality (3.3X) (30). Another analysis demonstrated that ALT>43 IU/l for men and >30 IU/l for women was related to the presence of coronary heart disease, even when patients with obesity, chronic viral hepatitis, and excessive alcohol use were excluded (31).
Impact of using a lower ALT ULN on clinical practice
Utilizing a significantly lower ULN for ALT will have implications by defining many more patients as having abnormal ALT levels. For example, applying the calculated “maximum correct classification” for ULN of ALT (29 IU/l for men and 22 IU/l for women) to the NHANES databases would result in 36% of men and 28% of women being defined as having elevated ALT (26). However, multiple studies from the NHANES database have demonstrated that indeed the prevalence of individuals with elevated ALT levels has increased significantly in the US, likely from the obesity epidemic and resultant NAFLD (32).
Some have argued against lowering of the ALT ULN due to major clinical and financial implications including increased health care costs and unnecessary evaluations, increased mental anguish and anxiety, and reducing the blood donation pool (33). Supporting these concerns was a study of 235 asymptomatic workers, in which 27% had abnormal liver tests (using lower ULN criteria), yet only six were found to have any liver disease on further evaluation although long-term mortality was not assessed in this study (34). Our belief, given the increased liver-related mortality demonstrated across multiple populations for ALT>33 IU/l for men and >25 IU/l for women, is that clinicians should be educated about the adverse long-term outcomes of these historically non-elevated levels and that a national effort should be undertaken to standardize ALT levels across all populations.
Further evidence for lowering the ULN of ALT and AST comes from reports demonstrating that significant liver disease may occur in the presence of “normal” liver chemistry levels. One report found that 9% of hepatitis C patients with normal ALT (defined as <50 IU/l) or near-normal ALT (defined as ≤1.4X ULN) had bridging fibrosis and 11% had cirrhosis (35). Another study treated hepatitis C patients with ALT levels <30 IU/l with peg interferon alfa-2a/ribavirin and noted further reduction in ALT levels by up to 10 IU/l ((ref. 36)). In a large cohort of patients with hemochromatosis, including 32% of cirrhotics, 40% had normal AST and ALT although the precise definition of normal ALT and AST levels was not reported (37). Another study demonstrated that an ALT cut off of 40 IU/ml was associated with a high prevalence of steatosis in a cohort where proton magnetic resonance spectroscopy was used to determine hepatic triglycerides as a marker of hepatic steatosis with 79% of those with elevated hepatic triglyceride levels having ALT<40 U/l in men and 31 U/l in women (38).
Previous guidelines have recommended basing clinical decisions to evaluate abnormal ALT levels based on multiples of the ULN of ALT (i.e., 2X ULN, 3X ULN, etc.) without specifically defining the ALT level (19, 20). This has led to variability in clinical practice due to the wide variation in the ULN of ALT across laboratories. For the purposes of this guideline, we have opted to define a “normal” ALT based on the available literature correlating ALT levels and liver-related mortality. However, clinical judgment still remains of paramount importance. If a patient has signs and/or symptoms of clinical liver disease, even in the absence of abnormal liver chemistries, an evaluation should be initiated. In addition, the linear relationship of ALT to BMI should also be considered when assessing for the presence of significant liver disease as studies have demonstrated that those with higher BMIs may have higher ALT levels and some have suggested that an ALT correction for being overweight should be considered (18, 39, 40).
Specific diseases of the liver including diagnostic testing
Viral hepatitis
Chronic hepatitis B and C infections are common in the United States. Approximately 4.1 million Americans are positive for antibodies against hepatitis C, and an estimated 3.0 million harbor chronic infection on the basis of positive hepatitis C RNA in the serum (41). The risk for acquiring hepatitis C is highest among individuals with parenteral exposure, such as from intravenous or intranasal drug use, blood transfusions before 1992, needle stick exposures, tattoos or body piercings, as well as high-risk sexual contact. The screening test for chronic hepatitis C is the hepatitis C antibody, which has a sensitivity of 92–97% (42). The positive predictive value of the antibody test is highest among individuals with risk factors for exposure; false positives may be observed in 5% of cases, and as high as 30% among individuals without reported risk factors. Confirmation of chronic infection is established by a highly sensitive HCV RNA PCR assay which has high sensitivity and specificity (AASLD/IDSA/IAS-USA. HCV testing and linkage to care. Recommendations for testing, managing, and treating hepatitis C (http://www.hcvguidelines.org/full-report/hcv-testing-and-linkage-care)). Individuals confirmed to have positive RNA should be referred to a specialist for further characterization of the infection and assessment of liver fibrosis to guide the decision to pursue antiviral therapy. As recommended by the Centers for Disease Control (CDC) and United States Preventive Services Task Force (USPSTF), individuals born within the 1945–1965 birth cohort should be considered for universal HCV antibody testing independent of abnormalities in AST/ALT levels, which may be normal in the presence of chronic infection including advanced liver disease (41, 43). Acute infection with hepatitis C is a rare presentation compared with chronic hepatitis C and is associated with higher levels of aminotransferase levels, but is typically anicteric and without clinical symptoms of hepatitis (44). The diagnosis of acute hepatitis C can be made in the setting of recent risk factors including drug use, and more recently men having sex with men has been identified as an increasingly prevalent risk factor. Testing includes anti-HCV, which is typically positive 6–8 weeks after exposure, with confirmation by measuring HCV RNA by PCR testing which must be done to confirm a case of acute hepatitis or chronic hepatitis C.
Testing for hepatitis B should be performed in all patients with persistently elevated AST/ALT levels. Chronic hepatitis B infects ˜1.5 million Americans and over 280 million individuals worldwide (23). The route of transmission is predominantly vertical or horizontal in Asia and Africa where hepatitis B is endemic, and predominantly through parenteral or sexual routes in the U.S. and other western nations. Characterization of an individual’s hepatitis B status can be achieved with three serologic tests, including the hepatitis B surface antigen (HBsAg) which is indicative of hepatitis B infection, the hepatitis B core antibody total which signals prior exposure, and the hepatitis B surface antibody, which signal immunity to the infection, either natural or vaccine-mediated (23). Chronic infection is confirmed by presence of the HBsAg and/or positive viremia on a highly sensitive HBV DNA assay. Individuals confirmed to have chronic hepatitis B should be referred to physicians with expertise in its management, and undergo further characterization of their infection with tests such as hepatitis B e antigen, hepatitis B e antibody, hepatitis B genotype, hepatitis B viral load (quantitative DNA), and fibrosis assessment, which may guide the decision for antiviral therapy. The diagnosis of acute hepatitis B is made by a positive immunoglobulin M (IgM) hepatitis B core antibody (hepatitis B core antibody IgM) and HBsAg in the setting of an acute hepatitis (45). Unlike acute hepatitis C in adults, acute hepatitis B infection is more commonly associated with signs and symptoms of hepatitis.
Non-alcoholic fatty liver disease
NAFLD is a highly prevalent condition associated with the metabolic syndrome, and is observed most commonly in patients with co-existing disorders including obesity, diabetes mellitus, dyslipidemia, and hypertension and should be strongly considered in individuals with mild elevations of AST/ALT levels (46, 47). Unlike alcoholic liver disease, there is no unique pattern of elevation for ALT and AST levels, although in general, ALT is higher than AST levels, and levels are rarely above 300 IU/l. Although one-third of Americans may meet a broad definition of NAFLD based on the presence of hepatic steatosis, a much smaller subset have NASH which is characterized by inflammation, fibrosis, and the potential for progressive to cirrhosis. No serological test is presently available to distinguish NAFLD from NASH or establish a diagnosis of NASH, and therefore it frequently represents a diagnosis of exclusion in which a series of diagnostic labs are obtained to first rule out alternative etiologies. Although many individuals with NAFLD suggested by steatosis on imaging may have normal liver chemistries, the presence of abnormal liver chemistries signals a higher likelihood for NASH with or without fibrosis and therefore warrants clinical evaluation. Compatible liver imaging for fatty liver infiltration on liver ultrasound, computed tomography, and magnetic resonance imaging (MRI) are useful to establish fatty liver itself, although liver biopsy is required to establish a diagnosis of NASH. As it is not practical to offer all patients with NAFLD a biopsy, guidelines have been established to help determine which NAFLD patients may require biopsy (46). Vibration-Controlled Transient Elastography (FibroScan device, Echosens, Paris, France) was recently approved by the Food and Drug Administration (FDA) in the US, and may be a useful non-invasive tool to determine who might have more advanced fibrosis and controlled attenuation parameter measurement may be incorporated with elastography to quantify steatosis although the role of the these non-invasive techniques is still emerging (48).
Alcoholic liver disease
Alcohol consumption is a very common cause for elevated liver chemistries, both independently, and as a contributor to liver injury in patients with other chronic liver diseases such as hepatitis C infection. As such, directed query and characterization of alcohol intake by patients is essential to identifying alcoholic liver disease as the precipitating factor. The definition of significant alcohol consumption has been suggested as >210 g of alcohol per week in men and >140 g per week in women (46). Radiographically and histologically, the pattern of liver injury is similar to that of NAFLD and these can overlap as dual contributors to acute and chronic liver disease. Specific patterns of liver injury have been commonly associated with alcoholic liver injury, including an AST:ALT ratio of at least 2:1, with values of AST or ALT rarely exceeding 300 IU/l (ref. 49). A higher ratio of AST:ALT exceeding 3:1 further increases the likelihood of alcoholic liver disease, and this ratio largely reflects the relatively lower serum activity of ALT in comparison to AST driven by pyridoxine deficiency observed in patients with alcoholic liver disease (50). Measurement of GGT may represent a complementary test to identify patterns of alcoholism or alcohol abuse, although GGT by itself is not helpful in establishing a diagnosis of alcoholic liver disease (51). Alcohol consumption should be queried in all patients presenting with abnormal liver chemistries, and complete cessation should be recommended in all patients, and in particular those patients for whom immediate serologic examination is deferred and close surveillance is planned.
Autoimmune liver disease
Chronic autoimmune hepatitis represents an important cause of chronic liver disease which is associated with persistently elevated liver chemistries, and may occur in the presence of other autoimmune disorders such as hypothyroidism, ulcerative colitis synovitis, Sjogren’s, rheumatoid arthritis, and psoriasis. Systemic lupus erythematosus is often mentioned in association with AIH, but the association is not strong (52). Typically observed more commonly in women than men (4:1), it has a lower prevalence than viral hepatitis or alcoholic/NAFLD (1:6,000), and is identified by the presence of characteristic serologic markers, including anti-nuclear antibody, anti-smooth antibody, and less commonly anti-liver kidney microsomal antibody or anti-soluble liver antigen antibody. The presence of hypergammaglobulinemia on testing for immunoglobulin G (IgG) or serum protein electrophoresis is suggestive although not diagnostic. A diagnosis of autoimmune hepatitis requires histologic confirmation on liver biopsy which may confirm a diagnosis, exclude an overlap syndrome with related autoimmune liver disorders, and stage for liver fibrosis.
Metabolic/genetic disorders
Hereditary hemochromatosis remains one of the most common inherited disorders which affects the liver, and hereditary hemochromatosis (HFE)-associated hemochromatosis particularly affects Caucasians of northern European origin. The prevalence of the HFE homozygous mutation C282Y is observed at a prevalence of 1 in 220–250 individuals with a heterozygote prevalence C282Y of 1 in 10 (ref. 53). The presence of a positive family history, or medical history/physical exam features suggestive of end-organ involvement of iron overload within the liver, pancreas, skin, joints, or heart, may all signal a probable diagnosis. However this presentation is rarely encountered as the penetrance or proportion of individuals with C282Y homozygous mutations who express these findings is quite low (54). Screening should be considered in all patients with abnormal AST/ALT levels, and include an iron panel from which transferrin saturation and serum ferritin can be determined. If the transferrin saturation is 45% or higher, or the serum ferritin is elevated, consideration should be given for HFE gene mutation analysis. It should be noted that ferritin is also an acute phase reactant and therefore an elevated ferritin in the setting of an acute hepatitis likely does not indicate iron overload. The presence of a homozygous C282Y mutation confirms HFE-related hereditary hemochromatosis; compound C282Y/H63D heterozygotes rarely manifest chronic liver disease from iron overload (55). In patients with either homozygous C282Y or compound heterozygous C282Y/H63D mutations who have elevated AST/ALT levels or ferritin >1,000 μg/l, or non-HFE hemochromatosis, liver biopsy should be considered to stage liver fibrosis, and quantify hepatic iron overload to guide therapy.
Wilson’s disease is a rare autosomal recessive disorder of biliary copper excretion due to a defective ATP7B copper transporter protein, which occurs in ˜1:30,000 individuals and represents an uncommon cause of abnormal AST/ALT levels. Although most commonly diagnosed in young males, it can present at any age. Wilson’s disease can present with hepatic, neurologic, and/or psychiatric manifestations. The liver presentation can be variable and may include hepatosplenomegaly, abnormal liver enzymes, cirrhosis, and acute liver failure. Screening should be considered in all patients with persistently elevated AST/ALT levels, and consists of serum ceruloplasmin testing (56). If low, confirmatory testing should be pursued including 24-h urine copper and/or serum copper levels, slit-lamp eye examination to identify pathognomonic Kayser-Fleischer rings, and possible liver biopsy to confirm the diagnosis, stage liver fibrosis, and quantify hepatic copper overload to guide management. Over 32 different mutations of the Wilsons gene have now been identified and unlike hemochromatosis, there is no predominant mutation. Molecular genetic analyses for the ATP7B mutation can be pursued in cases of diagnostic uncertainty.
Alpha-1 anti-trypsin deficiency is a rare genetic disease in adults, although it represents the most common genetic cause of liver disease in children, occurring at a prevalence of 1:2,500 in North American Caucasians. Defective production of the alpha-1 anti-trypsin protein may result in both panacinar emphysema and chronic obstructive pulmonary disease, as well as progressive liver disease, liver cirrhosis, and hepatocellular carcinoma. Screening should be considered in all patients with persistently abnormal liver AST/ALT levels, and consists of testing for low quantitative levels of lpha-1 anti-trypsin and genotype testing for the PiZZ mutation which results in severe enzyme deficiency (57, 58). Those with heterozygote phenotype (most commonly PiMZ) may also be at risk for liver disease, particularly in the presence of concomitant steatosis or viral hepatitis (59).
Drug and supplement-induced liver injury
Prescribed and over-the-counter medications, as well as non-prescribed complimentary alternative medicines or dietary supplements, represent a common source for acute and chronic liver injury. Identification of an offending agent is challenging and requires a comprehensive query of the patient, his or her family, and careful review of available medical and pharmacy records and laboratory data (22). Nearly all medications are associated with at least a small risk of elevation of ALT/AST/alkaline phosphatase or bilirubin with or without hepatotoxicity. Common drug classes that may cause elevated liver chemistries include antibiotics, antiepileptics, nonsteroidal anti-inflammatory agents, HMG-Co-A-reductase inhibitors (statins), anti-tuberculosis drugs, anti-retroviral treatment for HIV, biologic agents such as anti-tumor necrosis factor drugs, and some cancer chemotherapeutic agents. Of note, HMG-Co-A-reductase inhibitors (statins) have been associated with elevated ALT and AST levels but there is an extensive literature on the safety in those with chronic liver disease with only rare cases of hepatotoxicity reported (60, 61). In those with high ALT levels (>1,000 IU/l) an accurate history of acetaminophen use either as a single medicine or in combination with analgesic-narcotic combinations is essential. Attributing liver injury to a specific agent frequently requires empiric trials of drug discontinuation to observe full recovery of liver chemistries, although in cases in which the suspected agent is medically required, efforts should be made to identify potential drug alternatives, and/or establish a close surveillance plan to monitor for progressive liver injury or hepatic failure. In cases of acute or severe fulminant hepatitis, liver biopsy may be required to confirm severity of liver injury and establish a diagnosis of offending agent. Specific query for non-prescribed supplements is essential to discovery of associated liver injury. Common herbal supplements associated with hepatotoxicity include chaparral, ephedra, ji bu huan, germander, green tea extract, and shark cartilage (8, 62). A helpful resource that is available to clinicians trying to ascertain whether a drug or supplement may be hepatotoxic is the website livertox.nih.gov.
Patients with confirmed alkaline phosphatase elevations
Cholestatic liver disorders
Primary biliary cholangitis (PBC), formerly known as primary biliary cirrhosis, is an uncommon chronic liver disease primarily affecting intralobular bile ducts at the microscopic level (63). Although it is a rare cause of elevated liver tests, it is among the most common chronic cholestatic liver disorders and is seen more commonly in women than men. Patients may present with primary complaints of fatigue and pruritus. Lab testing reveals biochemical evidence of cholestasis (elevated alkaline phosphatase with or without elevation of bilirubin), as well as the serologic hallmark for screening and diagnosis: positive anti-mitochondrial antibodies which are observed in over 95% of patients (64). Liver biopsy is not routinely required although may be beneficial in patients with suspected anti-mitochondrial antibodies-negative disease and to stage liver fibrosis.
Primary sclerosing cholangitis (PSC) also represents a rare chronic liver disease which is characterized by chronic immune-mediated inflammation and fibrosis of both intrahepatic and extrahepatic bile ducts, fixed multifocal bile duct strictures, progressive liver fibrosis and cirrhosis, and an increased risk for both hepatocellular carcinoma and cholangiocarcinoma (65). Although no serologic hallmarks have been identified for PSC, IgG and anti-neutrophil cytoplasmic antibody are frequently elevated, and IgG4 may signal a specific IgG4-associated form of PSC. Diagnosis of PSC is established by characteristic imaging findings of thickened, inflamed bile ducts, dilatation of intrahepatic ducts, and focal strictures on MRI cholangiography or endoscopic retrograde cholangio-pancreatography. Although periductal concentric fibrosis or “onion-skin” like patterns of fibrosis are characteristic histologic findings, these signs are uncommonly seen and therefore liver biopsy is rarely required to establish a diagnosis or to significantly alter medical management. There is a common association with inflammatory bowel disease of the colon. Chronic cholestatic liver injury in the absence of PBC or PSC should prompt further investigation for less common etiologies such as bile duct obstructions, medications, sepsis, total parenteral nutrition, vanishing bile duct syndrome, and sarcoidosis (66).
Rare causes
A spectrum of non-hepatic disease may be associated with abnormal liver chemistries and referral to a hepatologist may be helpful in this circumstance. Celiac disease is a common disorder of gluten sensitivity which may be associated with modest elevations of liver transaminases; screening should be considered in patients with persistently elevated liver chemistries and consists of tissue transglutaminase IgA, and serum IgA level or tissue transglutaminase IgA and anti-deamidated gliadin peptide IgG (67, 68). Disorders of striated muscle stemming from inborn or acquired muscle disorders, or alternatively from muscle trauma or strenuous use for sport and exercise, may additionally cause abnormal AST/ALT levels with predominant involvement of AST (69). Screening may be considered after other more common etiologies have been ruled out, and initially consists of creatine kinase and/or aldolase levels. Both hypothyroidism and hyperthyroidism have been associated with abnormal liver enzymes, including both hepatocellular and cholestatic patterns of injury, particularly in more severe cases of myxedema and/or thyrotoxicosis. Screening should be considered in patients with compatible medical history and consist initially of thyroid stimulating hormone, and selective testing of free T4 and free/total T3. Liver disorders of pregnancy are uncommon but critically important conditions to identify in a time-sensitive fashion due to their potential effect on clinical outcomes for both mother and fetus. Trimester-specific consideration should be given for cholelithiasis, intrahepatic cholestasis of pregnancy, acute fatty liver of pregnancy, pre-eclampsia, and exacerbation of other chronic primary liver disorders (68). Biochemical patterns of liver injury, clinical history, and physical exam represent the primary guides to establishing a diagnosis, which may require consideration for early delivery in severe cases. Liver biopsy is rarely required to establish a diagnosis and guide management. In some regions of the U.S., tick-borne illnesses such as Lyme disease, babesiosis, and ehrlichiosis are endemic; these infectious are uncommonly associated with abnormal liver chemistries with predominant hepatocellular pattern (70, 71, 72). In patients with a compatible medical history, screening may be considered with specific testing for Borrelia burgdorferi, ehrlichiosis of human granulocyte anaplasmosis or human monocyte forms, or babesiosis microti.
Clinical assessment of the patient with abnormal liver chemistries
Summary statements:
Clinical assessment of the patient with elevated liver tests should begin with a thorough history and physical examination.
- History should include risk factors for underlying liver disease, associated medical conditions, use of alcohol, and use of medications including over-the-counter products and herbal supplements.
- Physical examination should assess for stigmata of chronic liver disease, as well as signs or symptoms pointing to a specific liver disease etiology.
Before initiation of evaluation of abnormal liver chemistries, one should repeat the lab panel and/or perform a clarifying test (e.g., GGT if serum alkaline phosphate is elevated) to confirm that the liver chemistry is actually abnormal. The initial history should assess risk factors for liver diseases including viral hepatitis, metabolic liver disease, exposures to toxins, and medicines including alcohol and complementary-alternative medicines, associated diseases including inflammatory bowel disease, hematologic diseases, pulmonary disease, and cardiac disease as well as hereditary forms of liver disease. In the patient who presents with jaundice, a history of blood disorders may suggest hemolysis. In those with intrahepatic cholestasis, light-colored stools, pruritus, and dark urine (bilirubinuria) may be elicited. The patient with hematologic diseases may have hepatic vein thrombosis presenting with acute pain in the right upper quadrant, ascites, hepatomegaly (73). A history of hemochromatosis may be suggested by a history of diabetes, skin pigmentation, cardiac disease, arthritis, and hypogonadism.
The risk factors for hepatitis B and C have been previously discussed. Hepatitis A and E cause acute hepatitis and are transmitted via fecal-oral route, and while acute hepatitis A is common, hepatitis E is uncommon in the United States although should be considered when there is a travel to an endemic area (Central America and Asia) (74). With acute viral hepatitis of any etiology, constitutional symptoms may also be present including nausea, vomiting, fever, and anorexia.
In those who present with jaundice and abdominal pain, obtaining a history of prior hepatobiliary disease including gallstones and inflammatory bowel disease is recommended. Other extrahepatic causes of abnormal liver tests include a history of heart failure (congestive hepatopathy), early onset emphysema, celiac disease, and thyroid disease. Finally, a family history of inherited liver disorders including hemochromatosis, Wilson disease, alpha-1-antitrypsin deficiency should be elicited.
The physical examination of a patient with abnormal liver tests is typically normal, but certain findings might help to confirm the presence of chronic liver disease including jaundice, ascites, splenomegaly, palmar erythema, spiders, and hepatic encephalopathy. Spider nevi, palmar erythema, gynecomastia, and caput medusae are typically signs of end-stage liver disease with portal hypertension and high estrogen levels (75). The presence of a palpable liver increases the likelihood of hepatomegaly being present but in and of itself does not imply an enlarged liver is present (76, 77). A firm liver edge due to cirrhosis or an infiltrative disorder is also more likely to be detected on exam (78). The presence of a palpable spleen and an enlarged left lobe of the liver suggest the presence of cirrhosis (78). Certain findings on physical examination may suggest specific etiologies of liver disease. Dupuytren contractures with parotid gland enlargement, and testicular atrophy may be seen in alcoholic liver disease (79). The presence of Kayser-Fleischer rings in patients presenting with neurologic symptoms and abnormal liver tests may be seen in Wilson disease. A bronzing of the skin may be seen in hereditary hemochromatosis. Marked hepatomegaly may be seen in acute viral hepatitis or alcoholic hepatitis with an enlarged nodular liver suggesting the presence of malignancy. Right upper quadrant tenderness with a positive Murphy sign may suggest hepatobiliary disease including cholecystitis. Markedly reduced breath sounds may be seen in alpha-1-antitrypsin deficiency with decreased breath sounds at either base suggesting a hepatic hydrothorax in the absence of clinically obvious ascites.
Patterns of liver chemistry test elevations
Summary statements:
- Hepatocellular injury is defined as disproportionate elevation of AST and ALT levels as compared with the alkaline phosphatase level.
- Cholestatic injury is defined as disproportionate elevation in alkaline phosphatase level as compared with AST and ALT levels.
- Mixed pattern of injury is defined as elevation of both alkaline phosphatase and AST/ALT levels.
- Isolated hyperbilirubinemia is defined as elevation of bilirubin with normal alkaline phosphatase and AST/ALT levels.
- The R ratio is calculated by the formula R=(ALT value÷ALT ULN)÷(alkaline phosphatase value÷alkaline phosphatase ULN) with an R ratio of >5 defined as hepatocellular injury, <2 cholestatic injury, and 2–5 mixed pattern.
The magnitude of the liver chemistry abnormalities and the ratio of AST to ALT help to guide the patient evaluation. Also, when examining the liver chemistry profile, the patterns of elevations are useful to suggest whether a patient’s liver test abnormalities are related to hepatocellular injury (elevated AST and ALT), cholestatic injury (elevated alkaline phosphatase), or a mixed pattern. An isolated hyperbilirubinemia may be seen in the presence of normal aminotransferase and alkaline phosphatase levels or may also accompany both hepatocellular and cholestatic disorders.
Several methods have been used to describe the pattern of abnormal liver chemistries. The most common classifications are (i) hepatocellular (disproportionate elevation in the AST and ALT levels as compared with the alkaline phosphatase level), (ii) cholestatic (disproportionate elevation in alkaline phosphatase level as compared with AST and ALT levels), (iii) mixed (elevation of alkaline phosphatase and AST/ALT levels), and (iv) isolated hyperbilirubinemia (elevation of bilirubin with normal alkaline phosphatase and AST/ALT levels) (80). Bilirubin levels also may be elevated in any of the first three categories. It should be noted that even though these patterns refer to the predominant enzyme elevation, the other liver tests may be elevated as well.
Recently, an R ratio has been used to assess whether the pattern of liver injury is hepatocellular, cholestatic, or mixed and may be applied in drug-induced liver injury (22). The R ratio is calculated by the formula R=(ALT value÷ALT ULN)÷(alkaline phosphatase value÷alkaline phosphatase ULN). An R ratio of >5 is defined as hepatocellular, <2 is cholestatic, and 2–5 is a mixed pattern.
Approach to evaluation for those with elevated AST and ALT
Summary statement:
- (Table 4 and Figures 1, 2, 3) A borderline AST and/or ALT elevation is defined as <2X ULN, a mild AST and/or ALT elevation as 2–5X ULN, moderate AST and/or ALT elevation 5–15X ULN, severe AST and/or ALT elevation >15X ULN, and massive AST and/or ALT >10,000 IU/l.
- Fulminant hepatic failure or acute liver failure, defined as the rapid development of acute liver injury with severe impairment of the synthetic function as manifested by prolonged prothrombin time and hepatic encephalopathy in a patient without obvious, previous liver disease requires immediate evaluation regardless of ALT level.

Causes of elevated AST and ALT

Algorithm for evaluation of aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) level. HCV, hepatitis C virus.

Evaluation of moderate elevation of aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels. HCV, hepatitis C virus.

Evaluation of severe elevation of aspartate aminotransferase (AST) and or alanine aminotransferase (ALT) levels. HCV, hepatitis C virus.
The magnitude of AST and ALT elevation varies depending on the cause of hepatocellular injury. Prior guidelines and studies have used a variety of definitions for AST and ALT elevations (19, 20). For the purposes of our guidelines, we will define borderline AST and ALT elevation as <2X ULN, mild AST and ALT elevations as 2–5X ULN, moderate AST and ALT elevations as 5–15X ULN, severe AST and ALT elevations as >15X ULN, and massive AST and ALT elevations as >10,000 IU/l.
Borderline and mild elevations of AST and/or ALT are seen in a variety of liver-related and non-liver-related conditions as shown in Table 4. Moderate elevations of AST and/or ALT overlap with the mild and severe lists. Severe elevations of AST and/or ALT can also come from a variety of sources including acute viral hepatitis, ischemic hepatitis/shock liver, septic shock, vascular disorders including acute Budd–Chiari syndrome or acute hepatic artery occlusion, toxin-/medication-induced liver injury, autoimmune hepatitis, acute biliary obstruction, diffuse infiltration of the liver with cancer, liver trauma/surgery, veno-occlusive disease/sinusoidal obstruction syndrome, HELLP syndrome, and Wilson’s disease. Massive liver-related elevations in AST and/or ALT to >10,000 U/l are generally only seen with shock liver/ischemic hepatopathy, or drug-induced/toxic hepatitis. Also, non-liver-related conditions such as rhabdomyolysis and heat stroke can result in severe to massive AST elevations.
Patients with moderate, severe, and massive elevations of ALT and/or AST require immediate evaluation (81). Such patients should be evaluated for acute hepatitis including acute hepatitis A, B, C, D, or E, acute reactivation of chronic hepatitis B, and acute hepatitis from non-hepatotropic viruses including herpes simplex, Epstein–Barr virus, or Cytomegalovirus. Acute autoimmune liver disease should also be suspected as well as idiosyncratic drug reactions or direct hepatotoxin exposure (acetaminophen). The highest levels of aminotransferase elevations are typically seen in those with acetaminophen overdose, ischemic hepatopathy, or toxin exposure, such as Amanita phalloides. A history of hematologic malignancy with recent chemotherapy may suggest a diagnosis of sinusoidal obstruction syndrome (veno-occlusive disease). And, a history of a hypercoagulable state in the setting of abnormal liver tests and ascites should suggest Budd–Chiari syndrome.
In addition to the magnitude of AST and ALT elevations, the ratio of AST to ALT may be useful in determining the etiology of abnormal liver tests (Table 4). Typically, for most liver conditions including chronic viral hepatitis and NAFLD, ALT levels are>AST levels. However, ˜90% of patients with alcoholic liver disease have AST>ALT, and >70% have an AST/ALT ratio>2 (82, 83). AST>ALT can also be seen in patients with cirrhosis of any etiology, although the AST:ALT ratio is typically not >2:1. In one study examining hepatitis C virus (HCV) patients, non-cirrhotics had an AST/ALT ratio of 0.60, while the mean ratio in cirrhotics was 1.05 (ref. 84).
A liver biopsy is most commonly done to assess the grade and stage of disease severity (hepatitis B, hepatitis C, and nonalcoholic fatty liver disease). In addition, biopsy may be useful to confirm a suspected specific diagnosis including Wilson’s disease (quantitative copper), hemochromatosis (hepatic iron index), or alpha-1-antitrypsin deficiency (periodic acid–Schiff-positive globules) (85).
Elevation of alkaline phosphatase level
Recommendations:
- (Table 5 and Figure 4) Right upper quadrant ultrasound should be performed in the setting of an elevation of alkaline phosphatase; if normal, evaluation for intrahepatic causes should be considered, including PBC, PSC, and drug-induced liver injury. (Strong recommendation, very low level of evidence).

Causes of elevated alkaline phosphatase

Continued.

Algorithm for evaluation of elevated serum alkaline phosphatase.
Cholestatic liver diseases are associated with elevated alkaline phosphatase, with or without elevated bilirubin. They can be categorized as either anatomic obstructions to bile flow (extra-hepatic cholestasis) or as functional impairments of bile formation by the hepatocytes (intra-hepatic cholestasis). Levels of alkaline phosphatase are also physiologically higher in children from rapid bone growth, and in pregnancy due to the production of placental alkaline phosphatase (86).
Determining the specific etiology of an elevated alkaline phosphatase level can generally be accomplished without the need for liver biopsy. In one study, ˜80% of isolated alkaline phosphatase elevations could be given a clinical diagnosis solely based on history and physical examination, routine labs, and chest X-ray in hospitalized patients (87).
If the alkaline phosphatase is elevated in the presence of other elevated liver chemistries, confirmation of hepatic origin is not required. With isolated alkaline phosphatase elevation, confirmation with GGT, or fractionation of alkaline phosphatase isoenzymes can be used to help differentiate liver alkaline phosphatase from non-liver sources. However, GGT elevation is not specific for cholestatic liver disease, and can be elevated in >50% of alcoholic patients without obvious evidence of liver disease (88, 89, 90, 91, 92). GGT can also be elevated in patients with pancreatic disease, myocardial infarction, renal failure, emphysema, diabetes, and in patients taking certain medications such as phenytoin and barbiturates (90, 93). Given its lack of specificity for liver disease, GGT should not be used as a screening test for underlying liver disease in the absence of abnormal liver chemistries.
Once alkaline phosphatase has been confirmed to be of hepatic origin, an ultrasound of the liver should be performed to assess the hepatic parenchyma and bile ducts. Biliary dilatation suggests an extrahepatic cause. A non-dilated biliary system suggests that the cause of elevated alkaline phosphatase is intrahepatic. Extrahepatic causes of cholestasis include choledocholithiasis, and obstruction of the biliary tract due to malignancy. Some disease of extrahepatic cholestasis may not be apparent by ultrasound including PSC or HIV/AIDS cholangiopathy and if these disorders are suspected such as in the context of a history of cholangitis or a history of colonic inflammatory bowel disease, cholangiography by MRI/magnetic resonance cholangiopancreatography (MRCP) or endoscopic retrograde cholangio-pancreatography should be undertaken.
For intrahepatic cholestasis, autoimmune markers including antimitochondrial antibody, antinuclear antibody, and smooth muscle antibody should be checked to assess for PBC or autoimmune cholangiopathy. A MRI/MRCP, endoscopic retrograde cholangio-pancreatography and/or endoscopic ultrasound can be ordered to better examine the bile duct morphology. Finally, pregnancy testing in women of childbearing age should be done to assess for intrahepatic cholestasis of pregnancy. Other infiltrative disorders may raise the alkaline phosphatase and cause intrahepatic cholestasis including sarcoidosis, atypical fungal infection, or malignancies. A liver biopsy may be considered to assess for primary biliary cirrhosis or other infiltrative diseases.
Elevation of total bilirubin level
Summary statement:
- (Table 6 and Figure 5) Elevated serum total bilirubin levels should be fractionated to direct and indirect bilirubin.
- An elevated serum conjugated bilirubin implies hepatocellular disease or biliary obstruction in most settings.

Causes of elevated bilirubin

Continued.

Algorithm for evaluation of elevated serum total bilirubin.
The first step in investigating elevated bilirubin is determining conjugated (direct) vs. unconjugated (indirect) hyperbilirubinemia. Elevated unconjugated or indirect bilirubin is due to over-production of bilirubin (such as hemolysis), decreased hepatic uptake, or decreased hepatic conjugation. The most common cause of elevated unconjugated bilirubin is Gilbert’s syndrome, affecting 3–7% of the US population (http://ghr.nlm.nih.gov/condition/gilbert-syndrome) which is due to a genetic defect of UDP-glucuronyltranferase resulting in decreased hepatic conjugation of bilirubin. Total bilirubin levels almost never exceed 6 mg/dl and are usually <3 mg/dl (94).
Fasting or significant illness can increase the unconjugated bilirubin by 2- to 3-fold, that in turn, will decrease with eating or administration of phenobarbital.
An isolated indirect hyperbilirubinemia may also be seen in the setting of hemolysis where reduced serum haptoglobin and elevated reticulocyte count and lactate dehydrogenase (LDH) level may be present. However, hemolysis infrequently causes a bilirubin level >5 mg/dl, unless co-existent renal disease, liver disease, or severe acute hemolysis is present (95). In general, in asymptomatic, healthy individuals who have mild unconjugated hyperbilirubinemia (<4 mg/dl), evaluation should exclude medications that cause hyperbilirubinemia, exclude evidence of hemolysis, and confirm normal serum transaminases and alkaline phosphatase levels. If these are found, then a presumptive diagnosis of Gilbert’s syndrome can be made and additional evaluation is not routinely necessary.
In contrast to indirect hyperbilirubinemia, conjugated (direct) hyperbilirubinemia generally implies the presence of parenchymal liver damage or biliary obstruction. This is associated with a number of hepatic disorders which result in decreased excretion of bilirubin into the bile ductules, and leakage of bilirubin from hepatocytes into serum. Total serum bilirubin levels may exceed 30 mg/dl in cases of severe liver damage, including alcoholic hepatitis with cirrhosis, and may be seen in advanced cirrhosis patients with sepsis and/or renal failure (51, 96, 97). In addition, an isolated hyperbilirubinemia may be seen after major surgery and typically resolves (98). The approach to elevated bilirubin is shown in Figure 5.
There are two rare inherited conditions associated with direct hyperbilirubinemia: Dubin–Johnson syndrome and Rotor syndrome where the direct bilirubin is ˜50% of the total with all other liver tests being normal including alkaline phosphatase and GGT levels. It is not necessary to distinguish the two disorders. Dubin-Johnson syndrome occurs due to a defect in the multidrug resistance canalicular enzyme while Rotor syndrome appears to be related to defective bilirubin storage by hepatocytes (99). Causes of elevated bilirubin are listed in Table 6.
In summary, clinicians often encounter elevated liver chemistries in practice. The evaluation of elevations of AST and ALT levels are guided by the clinical presentation and the degree of elevation. A normal ALT should be <29–33 IU/l in males and 19–25 IU/l in females. Minimal elevations may be approached by a history and examination, discontinuation of hepatotoxic medicines and all alcohol consumption, in addition to an evaluation for hemochromatosis, fatty liver and viral hepatitis. An evaluation for autoimmune liver disease, Wilson’s disease, and alpha-1antitrypsin deficiency is also warranted if the above tests are normal. Higher degrees of elevation (5–15 times the upper limit is normal) should be approached with an evaluation for viral hepatitis A, B, and C, iron panel, ceruloplasmin, alpha-1 antitrypsin phenotype, and autoimmune markers. If there are signs of acute liver failure, urgent hepatology consultation with referral to a liver transplant center should be undertaken immediately. Those who present with an elevation in alkaline phosphatase with normal AST, ALT, and bilirubin levels should have their alkaline phosphatase elevation confirmed with a GGT level and if elevated an ultrasound of the liver should be ordered. If the imaging is normal, autoimmune markers should be ordered. A liver biopsy may ultimately be required to obtain a diagnosis. An elevated bilirubin should be evaluated by determining conjugated and unconjugated fractions of bilirubin. An elevated conjugated bilirubin implies parenchymal liver damage or biliary obstruction. Extrahepatic causes such as hemolysis should also be assessed. Right upper quadrant ultrasound should be performed in patients with elevated conjugated bilirubin and alkaline phosphatase to assess for ductal dilatation. Finally, a liver biopsy may be required for diagnostic confirmation.
ACKNOWLEDGMENTS
This guideline was produced in collaboration with the Practice Parameters Committee of the American College of Gastroenterology. The Committee gives special thanks to Fred Poordad, MD, who served as guideline monitor for this document.
REFERENCES