MISATO: A Machine Learning Dataset of Protein-Ligand Complexes for Structure-based Drug Discovery
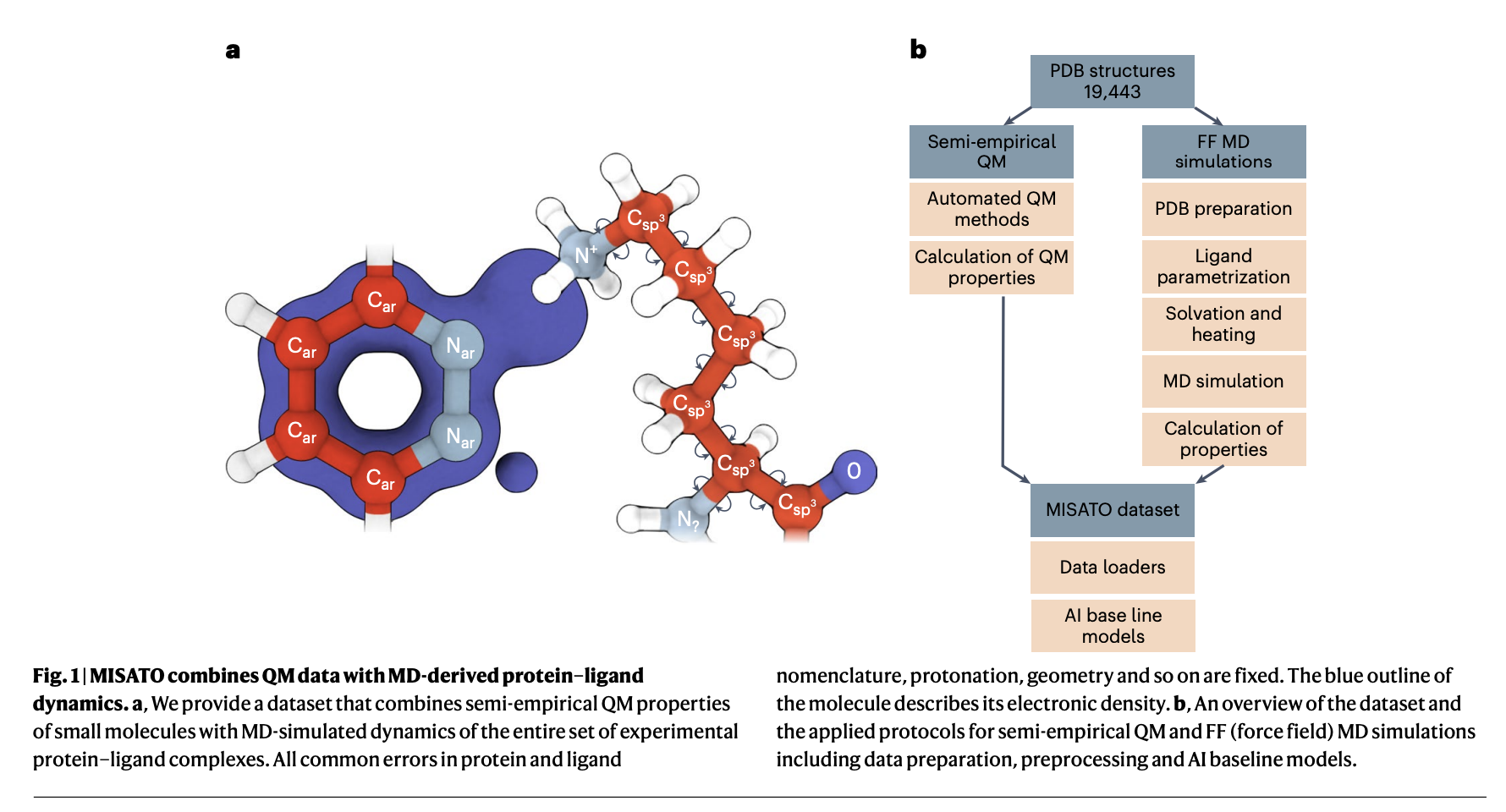
In the dynamic field of AI technology, a pressing challenge for the drug discovery (DD) community, especially in structural biology and computational chemistry, is the creation of innovative models finely tuned for drug design. The core challenge lies in accurately and efficiently predicting molecular properties crucial for understanding protein-ligand interactions and optimizing binding affinities, essential for advancing effective drug development initiatives.
In current structural biology and drug design, researchers commonly depend on existing datasets and methods, which have inherent limitations like structural inaccuracies, crystallographic artifacts, and difficulties in accurately capturing the dynamic nature of protein-ligand interactions. Traditional approaches for predicting molecular properties often lack the necessary detail for complex protein-ligand interactions, neglecting the vital role of dynamics and flexibility in understanding binding mechanisms and affinity.
Researchers from the Institute of Structural Biology, Technical University of Munich, Jülich Supercomputing Centre, Helmholtz AI, Cambridge University, Jagiellonian University, and Institute of Computational Biology propose MISATO, marking a transformative shift in drug discovery and structural biology methodologies. MISATO addresses the limitations of existing methods by integrating quantum-chemically refined ligand data, molecular dynamics (MD) simulations, and advanced AI models. This comprehensive approach facilitates a nuanced understanding of molecular properties, capturing electronic structure details and dynamic behavior crucial for accurate predictions.
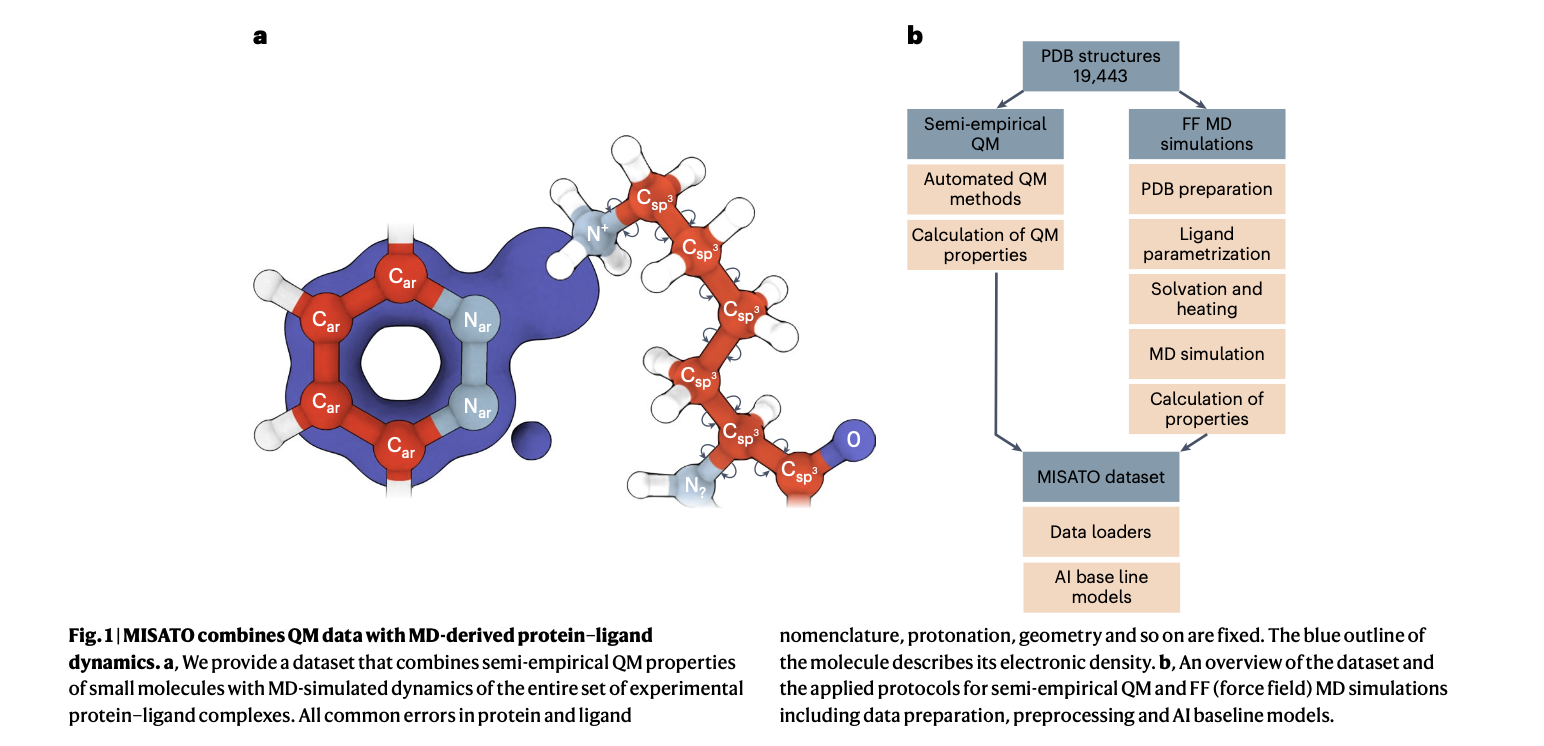
MISATO takes a comprehensive approach, utilizing semi-empirical quantum chemical methods to refine ligand datasets. This method captures electronic properties with high accuracy, while also analyzing both electronic structure details and dynamic behavior, crucial for precise predictions. Additionally, classical MD simulations within MISATO characterize the dynamic behavior and conformational landscape of protein-ligand complexes, offering insights into binding mechanisms and flexibility. AI models integrated into MISATO, such as graph neural networks (GNNs), are trained on this enriched dataset to predict properties like adaptability, binding affinities, and thermodynamic parameters. Extensive experimental validations confirm the efficacy of these models in accurately predicting key molecular properties crucial for drug discovery.
In conclusion, MISATO signifies a key stride in AI-driven drug discovery and structural biology. By integrating quantum chemistry, MD simulations, and advanced AI models, MISATO provides a holistic and robust solution to challenges in structure-based drug design, enhancing accuracy and efficiency and empowering researchers with potent tools.
Check out the Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter. Join our Telegram Channel, Discord Channel, and LinkedIn Group.
If you like our work, you will love our newsletter..
Don’t Forget to join our 42k+ ML SubReddit
Aswin AK is a consulting intern at MarkTechPost. He is pursuing his Dual Degree at the Indian Institute of Technology, Kharagpur. He is passionate about data science and machine learning, bringing a strong academic background and hands-on experience in solving real-life cross-domain challenges.
Discover more from reviewer4you.com
Subscribe to get the latest posts to your email.



